
Abstract
Cervicofacial vascular anomalies can result in morbidity, pain, and cosmetic concerns in affected individuals. Each anomaly has its own unique natural history, treatment, and associations with underlying genetic syndromes. For optimal patient care, it is important for the neuroradiologist to accurately recognize and characterize these entities to ensure appropriate treatment and management. In this review, we discuss the general characteristics, classifications, and imaging features associated with the most common vascular anomalies such as hemangiomas, arteriovenous malformations and fistulas, capillary malformations, venous malformations, and lymphatic malformations in the context of associated syndromes. Additionally, we discuss novel imaging techniques that aid in identifying these vascular anomalies.
Keywords
Background and classification
Cervicofacial vascular anomalies can result in morbidity, pain, and cosmetic concerns in afflicted individuals. It is important to accurately characterize, diagnose, and manage vascular anomalies in this anatomical region. Inconsistencies in the nomenclature of vascular malformations pose a challenge in uniformly classifying and managing vascular anomalies of the cervicofacial region. In response to these challenges, the International Society for the Study of Vascular Anomalies (ISSVA) has implemented a classification system based on extensive study of each distinct vascular anomaly; this schema was last revised in May 2018. 1
Vascular anomalies of the cervicofacial region can be subdivided into two categories: vascular tumors and vascular malformations. Vascular tumors can be benign, locally aggressive, or malignant.2,3 This review will cover primarily benign lesions. Vascular malformations, in contrast, result from defects in vascular morphogenesis, have quiescent endothelium and somatic growth, and can be divided into high-flow and low-flow lesions.2,4 High-flow vascular malformations, consisting primarily of arteriovenous malformations (AVMs) and arteriovenous fistulae, are pulsatile and under high pressure secondary to their arterial component. Low-flow vascular malformations, which include capillary malformations (CMs), venous malformations (VMs), and lymphatic malformations (LMs), are typically non-pulsatile due to the absence of an arterial component. There are also various combinations of the aforementioned malformations, as delineated by the ISSVA. 1 In this review, we aim to clarify differences among the most common vascular anomalies of the cervicofacial region and describe the classification and imaging features of these lesions in the context of the most commonly observed associated syndromes.
Benign vascular tumors and associated syndromes
Infantile and congenital hemangiomas
Classifications and characteristics
Hemangiomas are benign neoplasms characterized by endothelial proliferation. 2 The most common types of hemangiomas are infantile and congenital and are therefore primarily relevant to children. Other varieties include spindle cell and epithelioid hemangiomas. 3
Infantile hemangiomas consist of proliferating lobules of endothelial cells, which stain positive for GLUT1, and are the most common tumors of infancy.5,6 In 60% of cases, infantile hemangiomas occur in the head and neck.7,8 The incidence of head and neck infantile hemangiomas has been estimated to be ∼1% of infants. 9 Infantile hemangiomas may be absent at birth or visible as a small pallid or telangiectatic skin patch. A rapid growth phase follows for several months. A proliferation of feeding and draining vessels during this time is related to a high expression of angiogenic factors. After a brief growth plateau, the involution phase begins around 1 year of age. Lesion color fades as the size and thickness of the tumor decreases and eventually, it is replaced by fibrofatty tissue. While most infantile hemangiomas resolve by age five, resolution of deeper lesions may take up to seven or eight years.
Congenital hemangiomas are also characterized by lobular endothelial proliferation, but are distinct from infantile hemangiomas in that they are fully formed at birth, are GLUT1-negative, and are uncommon. 2 Congenital hemangiomas can be categorized into three subtypes based on the pattern of involution: rapidly involuting, partially involuting, and non-involuting.10,11 Somatic activating mutations in GNAQ and GNA11 were found in a sample of eight participants with various types of congenital hemangiomas. 12 Most infants with the rapidly involuting subtype demonstrate complete involution of the lesion in the first 6–14 months of life.10,13 As their names suggest, partially involuting types regress incompletely and non-involuting congenital hemangiomas remain present throughout life and typically require surgical excision if removal is desired. 10 It has been proposed that partially involuting congenital hemangiomas develop from incompletely involuted rapid-involuting subtypes to persist as non-involuting-like lesions. 14
Imaging features
Most hemangiomas are small and are diagnosed clinically. However, when the clinical findings are ambiguous, imaging may help delineate hemangiomas from other vascular malformations including venous and AVMs. 15 Hemangiomas can be diagnosed by ultrasound, especially when they are superficially located. In the proliferative phase, prominent vessels within the echogenic mass will have low arterial resistance and pulsatile venous waveforms on color Doppler evaluation.16,17 On magnetic resonance imaging (MRI), hemangiomas are hyperintense on T2-weighted images (T2WIs), show prominent flow voids, early intense enhancement and lack of peripheral edema.7,17 Time-resolved dynamic MR angiography (MRA) techniques help identify arterial feeders and draining veins. 18 Both MRI and digital subtraction angiography (DSA) can confirm the absence of arteriovenous shunting (Figure 1).19,20 Congenital hemangiomas can differ from infantile hemangiomas angiographically by demonstrating heterogeneous parenchymal staining, more prominent vascularity with large/irregular feeding arteries in disorganized patterns, minor direct arteriovenous shunting, and intravascular thrombi. 21 On ultrasound scanning (US), these differences can be evident as relative heterogeneous echogenicity and more visible vascularity. On magnetic resonance (MR) or computed tomography (CT), congenital hemangiomas may have more ill-defined margins and occasional adjacent fat stranding.19,20 Arterial spin-labeling typically shows marked hyperperfusion in congenital hemangiomas and infantile hemangiomas in the proliferative phase, which distinguishes these lesions from low-flow vascular malformations. 22
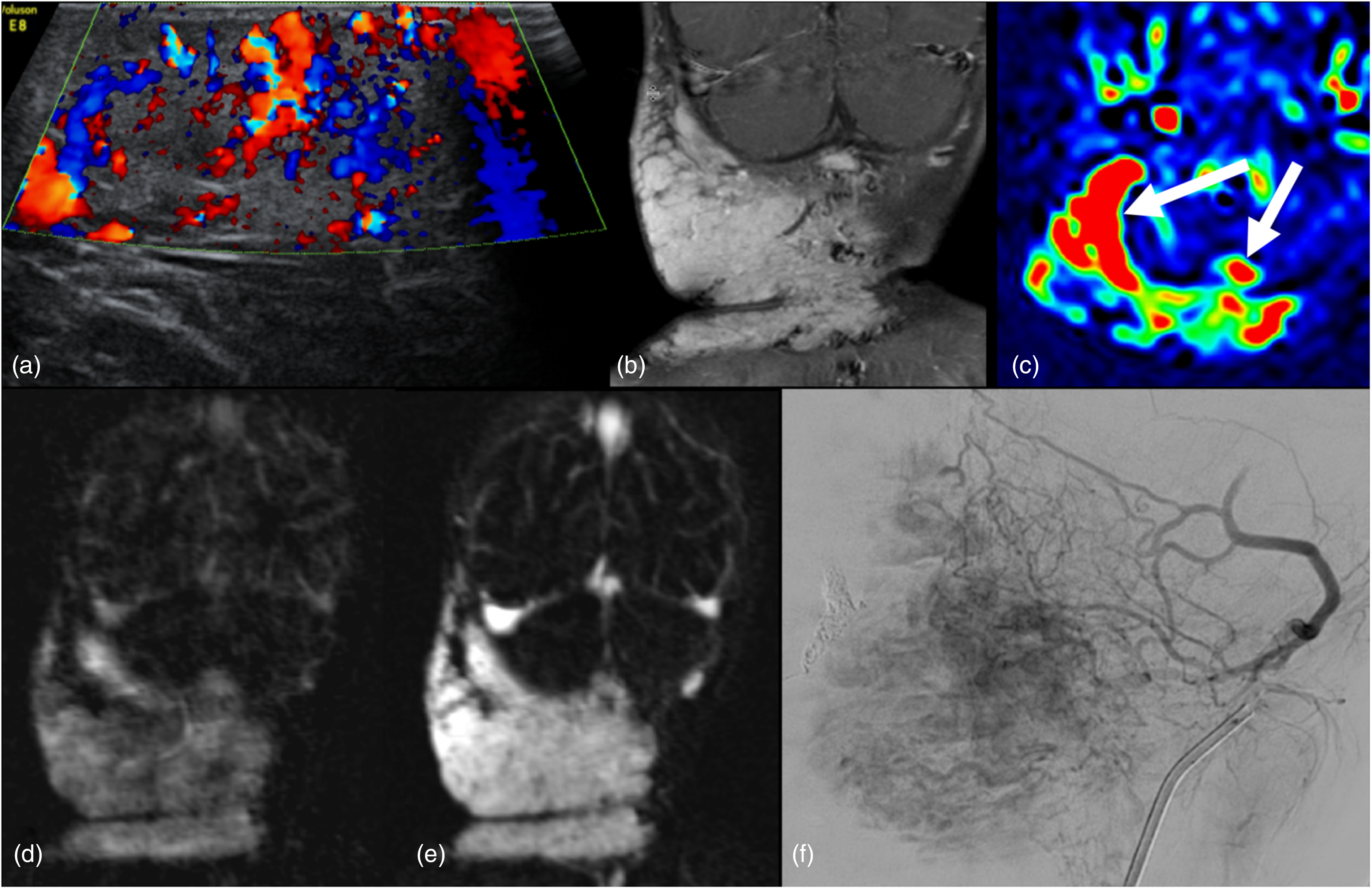
Infantile hemangioma. A 15-month-old girl presented with a large right neck mass that had been rapidly growing since 2 months of age. (a) Color Doppler ultrasound image of a large infantile hemangioma reveals prominent intralesional vascularity. (b) Coronal T1-weighted post-gadolinium better shows the extent of the avidly enhancing and lobulated mass centered in the right and posterior neck. (c) Axial ASL color map demonstrates intense flow-related signal within the lesion from focally elevated perfusion (arrows). Time-resolved MR angiography demonstrates progressive tumoral enhancement throughout arterial (d) and venous (e) phases. Digital subtraction angiogram (f) demonstrates hypervascular lobules of the hemangioma. ASL: arterial spin labeling; MR: magnetic resonance.
Expectant waiting is the initial approach for most hemangiomas, particularly for rapidly involuting congenital hemangioma and many infantile hemangiomas. Treatment is indicated for compromise of vital structures or skin ulceration. Propranolol is a highly effective treatment for infantile hemangioma that has replaced corticosteroids as primary medical therapy. Pulsed dye laser can be employed for ulcerated lesions. Excision with possible preoperative embolization is the treatment of choice for non-involuting congenital hemangiomas. 14 Surgery for infantile hemangioma is usually limited to excision of fibrofatty residua. 23
Associated syndromes
PHACES syndrome
PHACES syndrome is the most common syndrome observed in the context of cervicofacial hemangiomas. PHACES syndrome is a neuronal migration disorder that results in posterior fossa abnormalities, infantile hemangiomas in the face, head or neck regions, arterial lesions, cardiac anomalies, eye anomalies, sternal clefting, and/or a supraumbilical raphe. 24 Most hemangiomas in PHACES syndrome are segmental hemangiomas, which are large and plaque-like with a broad cutaneous distribution. 24 Segmental hemangiomas are at higher risk for complications such as bleeding, ulceration, visual compromise, airway obstruction, and auditory canal obstruction.24,25 Most of these complications occur in the proliferative phase. 26 Hemangiomas are also observed outside of the head and neck region in lumbar or pelvis syndrome and Maffucci syndrome.27,28
High-flow vascular malformations and associated syndromes
Arteriovenous malformations and arteriovenous fistulae
Classifications and characteristics
High-flow vascular malformations are rare in the cervicofacial region. AVMs consist of an anomalous artery to vein connection with an intervening nidus, but without an intervening capillary bed. Multiple feeding arteries and draining veins may be present. Most patients with cervicofacial AVMs present in childhood with facial swelling and erythema and occasionally with pain, mucosal ulceration, and bleeding.29,30 A palpable thrill and/or bruit over the lesion may be evident on physical examination. Most cervicofacial AVMs are located in the mid-face. In contrast to other vascular anomalies, AVMs typically have a progressive course that usually warrants intervention. 31
Arteriovenous fistulas are high-flow vascular malformations with a direct connection between an artery and a vein without an intervening nidus. 32 Cervicofacial arteriovenous fistulae are usually congenital or post-traumatic in origin,32,33 often presenting with pulsatile tinnitus or a pulsatile facial mass.32,34,35
Imaging
On ultrasonography, AVMs show a hypervascular network with multiple feeding arteries and draining veins, similar to infantile hemangiomas. Aside from not presenting as a discrete mass lesion, a distinctive feature of AVMs is the presence of arterial waveforms within venous structures due to arteriovenous shunting. 36 MRI typically reveals a tangle of enlarged vascular channels and flow voids.19,36,37 CT, CT angiography, and MRA better define vascular contributions; time-resolved techniques can help identify arterial and venous components and flow direction. 37 Regional changes can include fatty hypertrophy, edema, skin ulceration, and hyperostosis.36,38,39 Catheter-based DSA remains the gold standard for mapping and treating cervicofacial AVMs. Angiographic features include dilated arterial inflow into an intervening nidus, which can vary in size and configuration, and rapid shunting into dilated venous outflow tracts.36,40 In contrast to AVMs, arteriovenous fistulae demonstrate a large hypertrophied artery with a direct connection to a draining vein without an intervening nidus (Figure 2).36,40 Multiple feeding arteries may be present and the reflux of high-pressure arterial blood may cause multiple venous structures to become dilated with increased pressure. 36
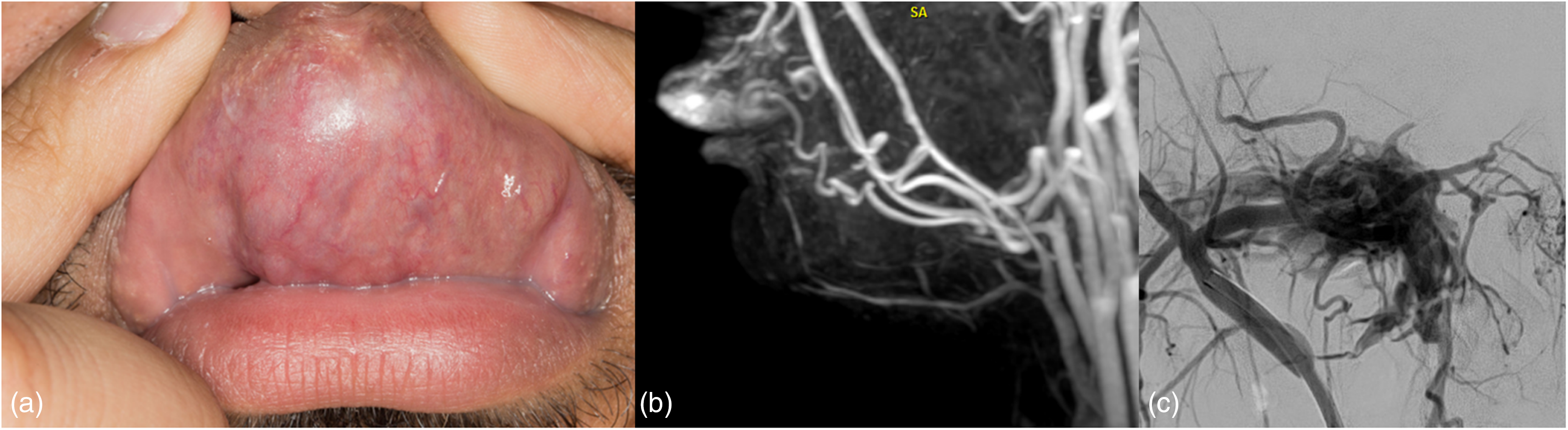
Facial arteriovenous fistula. (a) Photograph of the upper lip shows focal enlargement and increased vascularity from underlying malformation. (b) Sagittal MRA image better delineates dominant vascular components and the associated hypertrophy of the upper lip. (c) DSA of the internal maxillary artery showing a direct connection with the pterygoid venous plexus. DSA: digital subtraction angiography; MRA: magnetic resonance angiography.
In cases where treatment is pursued, post-procedural imaging is important in order confirm obliteration of the high-flow malformation. The follow-up modality utilized as well as imaging interval is institution-dependent. Furthermore, post-procedural imaging features will likely depend upon the treatment modality utilized. However, some general findings should be seen among successfully treated high-flow malformations. Regarding cervicofacial AVMs, evaluating for residual nidal tissue and evidence of arteriovenous shunting is important on follow-up imaging. Post-procedural DSA or time-resolved MRA will demonstrate no residual filling of the nidus and no evidence of early venous filling. Though less sensitive in detecting recurrent lesions than catheter-based DSA, MR-based evaluation offers a feasible, non-invasive method of follow-up evaluation. 41 In general, MR should demonstrate the diminished size of the nidus (or complete removal in cases of surgical resection) along with the resolution of dilated vascular flow voids.38,42 Regarding AVFs, DSA should demonstrate obliteration of the fistulous connection between the arterial and venous system along with the resolution of early venous phase opacification in the area of the fistula.
High-flow vascular malformations and associated syndromes
Cerebrofacial arteriovenous metameric syndromes
AVMs of the head and neck can be seen in the context of cerebrofacial arteriovenous metameric syndromes (CAMS). 43 In embryonic development, mesodermal, and neural crest cells that arise from the same metameric level migrate transversely to inhabit similar regions of the head and face. This includes embryonic tissues designated to develop into blood vessels. If an insult occurs prior to or during neural crest migration, vascular malformations can form within the same metameric segment.44,45 The resulting malformations can also be purely venous in nature; this is discussed hereafter. Three types of CAMS have been described based on metamere. CAMS 1 involves a defect in the medial prosencephalon with concurrent involvement of the hypothalamus and/or hypophysis, nose, and forehead. CAMS 2 arises from the lateral prosencephalon and involves the occipital lobe, optic pathway, thalamus, and maxilla. CAMS 3 derives from the rhombencephalon and manifests within the cerebellum, pons, and mandible. Symptomatic manifestations typically occur in childhood and can include visual defects, epistaxis, mucosal ulceration, facial asymmetry, and other cosmetic issues.
There is a wide spectrum of possible phenotypes in CAMS; one of these is Wyburn–Mason Syndrome, also known as Bonnet–Dechaume–Blanc syndrome. Malformations in Wyburn–Mason syndrome typically follow distributions of CAMS1 or CAMS2. Facial nevi or frank AVMs involving the maxillary or orbital regions are typically found with AVMs of the retina, optic pathway, midbrain, third ventricle, and thalamus. As these AVMs can rupture, patients are at risk for cerebral hemorrhage, epistaxis, and gingival bleeding.
Parkes Weber syndrome
Parkes Weber syndrome is an overgrowth disorder characterized by multiple small AVMs or AVFs associated with a cutaneous capillary stain as well as bony and soft-tissue hypertrophy of an affected limb.46,47 The phenotype of Parkes Weber syndrome is similar to that of Klippel–Trenaunay syndrome (KTS), however, the presence of high-flow vascular malformations in the former is the distinguishing feature between the two entities. 46 Parkes Weber syndrome is associated with mutations in the RASA1 gene, in contrast to KTS which is thought to be secondary to mutations in the PIK3CA gene, potentially accounting for their phenotypic differences.2,48,49
Low-flow vascular malformations and associated syndromes
Capillary malformations
Classification and characteristics
CMs consist of dilated capillaries and post-capillary venules within dermal tissue and are usually present at birth. 50 When CMs involve large portions of the face, they are referred to as port-wine stains or nevus flammeus. 51 Depending on their location, these lesions may darken or thicken with time. Isolated, non-syndromic CMs are associated with GNAQ mutations, as are the small portion associated with ocular and brain anomalies in the context of Sturge–Weber syndrome;39,52 these may be associated with soft-tissue or bony overgrowth. 39
Imaging
As CMs are superficial lesions, imaging is typically not required for diagnosis unless imaging is performed to evaluate for an underlying syndrome or a deeper lesion. On MRI, CMs are bright on T2WIs and enhance (Figure 3). 53 MRI of the brain may identify a pial capillary VM in children with Sturge–Weber syndrome. 53
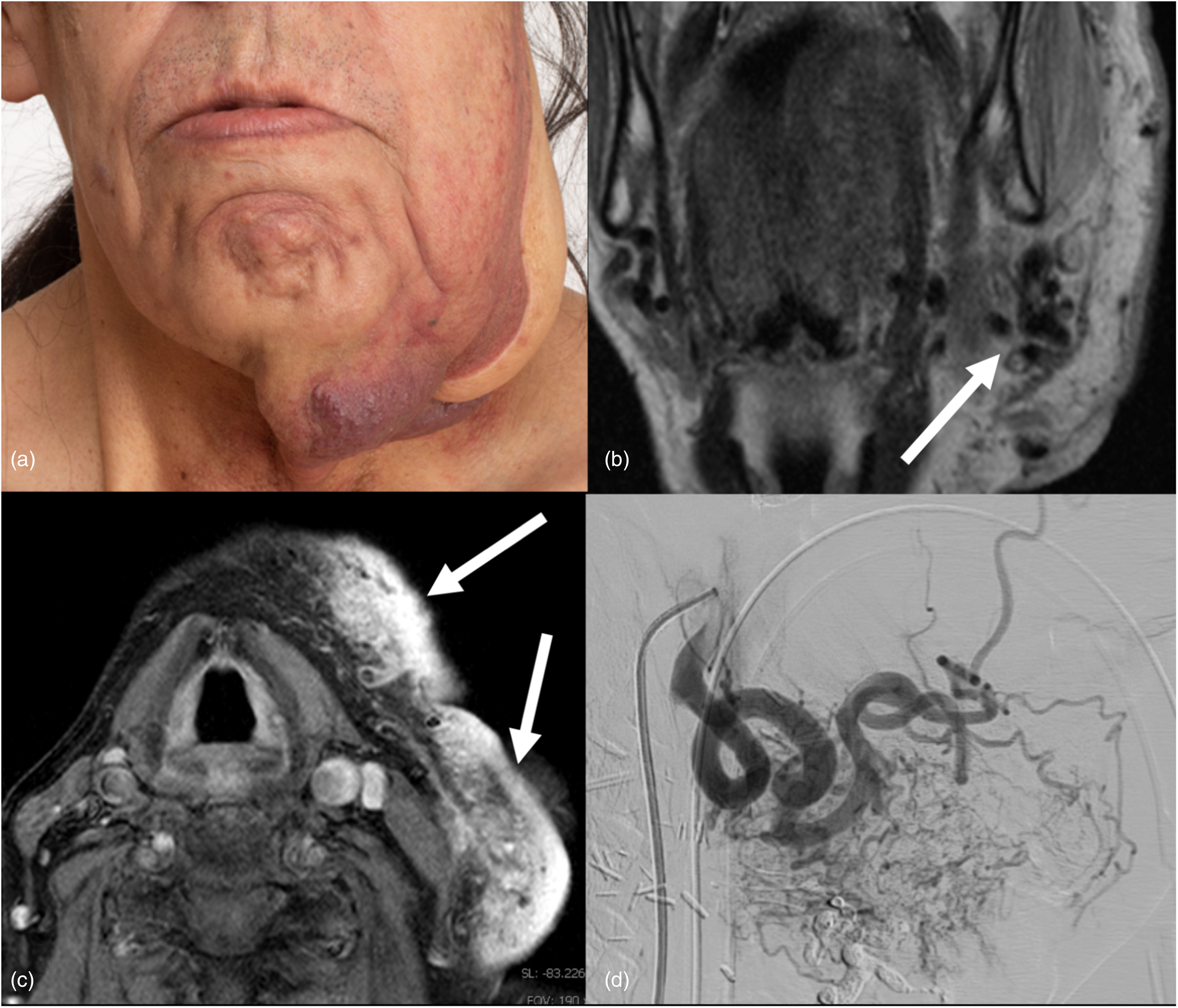
Arteriovenous malformation and concurrent capillary malformation. (a) Photograph of an extensive left facial capillary malformation. Facial asymmetry with enlargement of the left lower face is related to an underlying AVM. The patient has undergone prior partial facial resection with flap repair. (b) Coronal T2-weighted MRI demonstrates large flow voids indicating a high-flow vascular malformation (arrow). (c) Axial T1-weighted post-gadolinium fat-saturated image shows avid contrast enhancement of the capillary malformation at the level of the hyoid bone (arrows); involvement of cutaneous and subcutaneous tissues indicates possible additional venous components. (d) A sagittal view of a left external carotid artery cerebral angiogram shows a high-flow arteriovenous malformation of the left face which is supplied predominantly by the left facial artery. This was further treated with Onyx embolization. Genetic testing found a pathogenic variant in the gene RASA1; changes in this gene are responsible for CM–AVM syndrome and are consistent with his clinical features. CM–AVM: capillary malformation and arteriovenous malformation; MRI: magnetic resonance imaging.
Venous malformations
Classification
VMs are slow-flow, conglomerated venous networks consisting of dysplastic thin-walled veins that are GLUT1-negative. 54 Approximately 40% of VMs occur in the head and neck. 54 Of those located in the face, common locations include the lip, eyelid, cheek, and mucosal surfaces. 54 Osseous involvement can occur. 55 Patients with cervicofacial VMs typically present with a bluish discolored skin lesion that is soft to palpation and compressible.54,56 Children may complain of intermittent swelling and pain, and cosmetic concerns are a common reason for presentation.54,56 VMs increase in size in gravity-dependent positions and when they are subjected to increased venous pressure such as with the Valsalva maneuver. 54 Hemorrhagic complications in VMs are rare, although they may enlarge over time. 56
Imaging
Ultrasonography can be used in the initial evaluation of superficial VMs; on grayscale imaging VMs appear as heterogeneous hypoechoic masses with monophasic low-velocity flow on Doppler imaging.16,19 MRI remains a crucial modality in characterizing the extent of a VM and for treatment planning. 15 VMs are well defined and often transspatial. The involvement of critical structures may limit options for surgical resection. 15 VMs are markedly hypertense on fluid-sensitive MRI sequences, and show diffuse, gradual venous enhancement following contrast administration (Figure 4). 15 Phleboliths within a VM are pathognomonic but are not commonly seen; CT shows these as rounded calcifications and on MRI they are detected as foci of low signal on T2WIs or on susceptibility-weighted imaging.19,54 The typical treatment for VMs is percutaneous sclerotherapy. Follow-up imaging for VMs after sclerotherapy is typically performed with MRI, commonly including fat-suppression sequences. 57 Treatment efficacy is most commonly assessed by determining the reduction in size and extent of the VM following the intervention, which is best appreciated by the decrease in signal intensity volume on fat-suppressed T2WIs.57,58 This reduction in signal intensity is contributed by fibrotic scarring or thrombosis within the lesion following sclerotherapy. 59
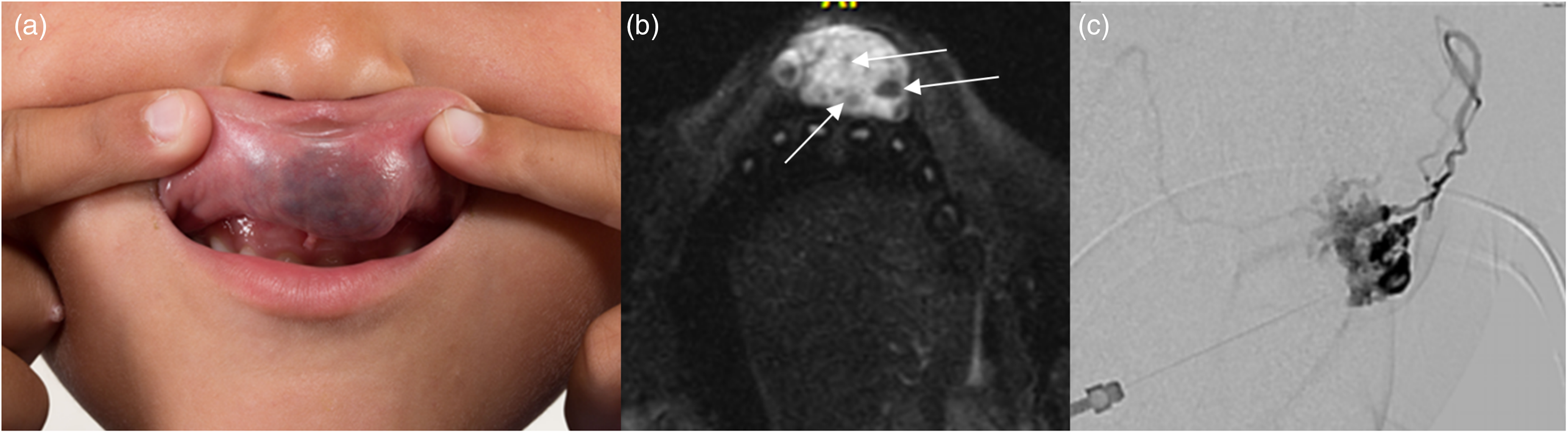
Venous malformation. (a) Photograph of the upper lip shows focal enlargement and blue discoloration from the underlying malformation. (b) Axial fat-saturated T2-weighted image shows a well-defined and avidly hypertense mass with intralesional hypointense foci consistent with phleboliths (arrows). (c) Direct puncture venogram demonstrates a slow-flow venous malformation.
Lymphatic malformations
Classification and characterization
LMs are benign congenital lesions that arise from embryological disruption of the developing lymphatic system. They present as multicystic, chyle-filled transspatial masses that are microcystic (<1 cm), macrocystic (>1 cm), or both.60,61 Anatomical areas with rich lymphatic supply are typically affected and approximately half of all cases involve the head and neck. 60 Children with LMs often present early in life and can be diagnosed prenatally by ultrasound or MRI, although presentation can occur at any age. 60 Most children come to clinical attention because of swelling and pain of the affected area, which is often related to enlargement of the lesion secondary to infection or intralesional hemorrhage. 62 Similar to VMs, LMs can enlarge over time. 62 LMs rarely regress spontaneously and therefore treatment involving medical management, sclerotherapy, and surgical excision may be indicated depending upon the severity of symptoms.61,63
Imaging
As with other low-flow vascular malformations, ultrasound provides first-line imaging for LMs while MRI better shows the complete extent. On ultrasound, LMs appear as a multilocular cystic mass with internal septae of varying thickness. 63 The cystic contents can show variable echogenicity but are typically hypoechoic.15,19 Lack of vascularity on color Doppler evaluation helps distinguish pure LMs from other vascular malformations, including mixed varieties.15,63 On MRI, quiescent LMs should have mostly fluid signal intensity. Internal debris and/or fluid–fluid levels, however, are commonly seen and indicate lesional inflammation or hemorrhage. 63 Macrocystic LMs can have faint peripheral or septal enhancement and microcystic lesions demonstrate robust enhancement due to the high density of internal septae (Figure 5). 63
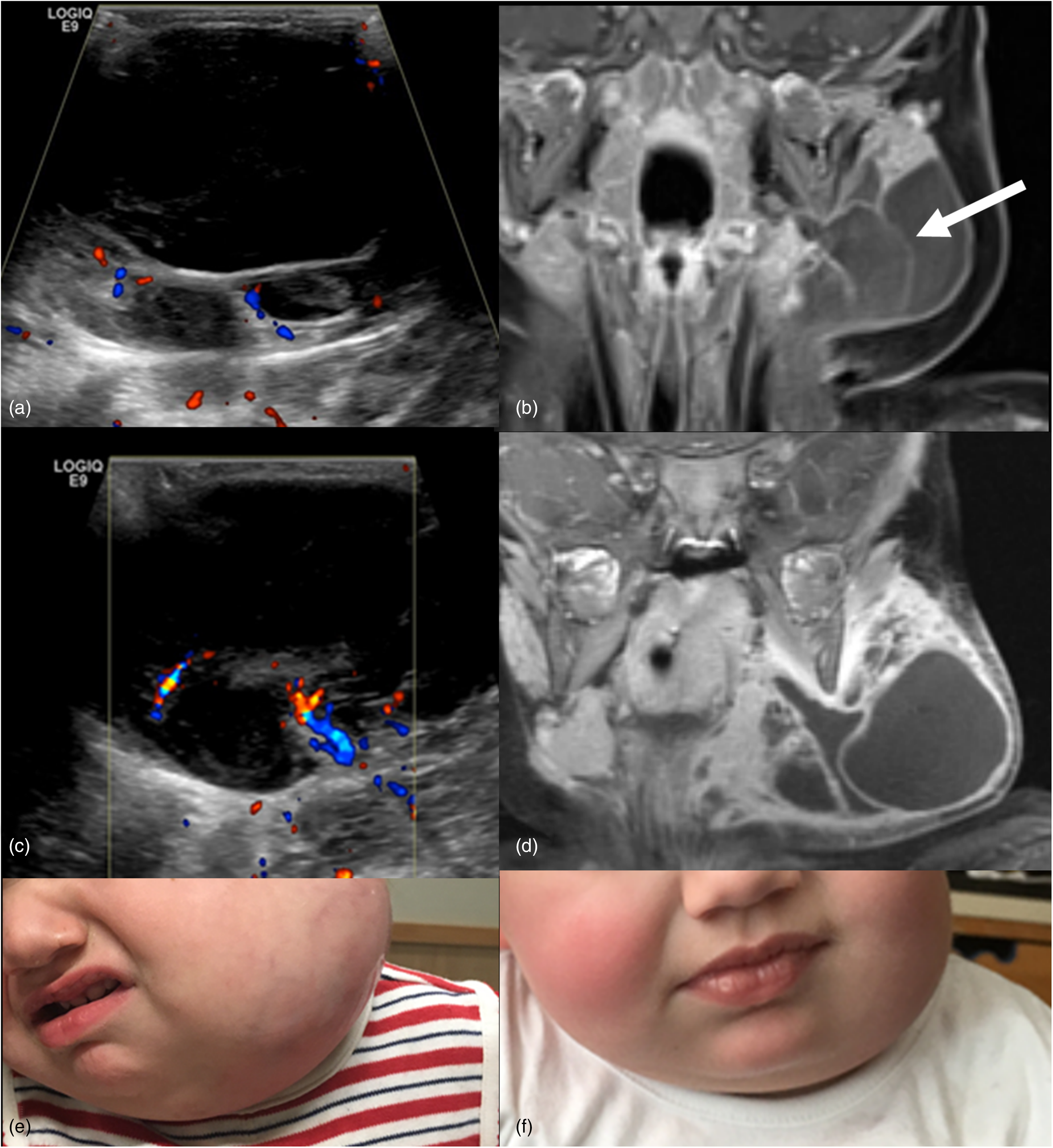
Lymphatic malformation. Two-year-old girl with a lymphatic malformation in the left face. Ultrasound image of the left face (a) shows a multicystic lesion with anechoic macrocysts and minor internal vascularity on Doppler evaluation. Coronal post-gadolinium T1-weighted (T1W) image (b) demonstrates a complex lymphatic malformation with a mild enhancement of the macrocyst septations (arrow). Patient returned 3 months later with a more acutely swollen and tender left face mass in the setting of fever. Follow-up ultrasound image (c) shows new scattered debris within the cystic components with more thickened and vascular septations. Coronal post-gadolinium T1W image obtained concurrently (d) shows marked expansion of the mass secondary to infection with avid enhancement of septations and microcystic components. Photographs of the lymphatic malformation during infection (e) and after infection (f) following antibiotic treatment.
A primary radiological outcome measure for successful non-surgical management of LMs (i.e. medical management or percutaneous sclerotherapy) is a reduction in size and extent of the malformation on US, CT or MR images, along with a resolution of mass effect on surrounding head and neck tissues.64,65 In addition to overall volume reduction, shrinking of both macro and microcystic components may also be observed, along with fibrous change within and surrounding the lesion as evidenced by low signal on T2WI sequences. 66
Syndromes with low-flow vascular malformations
Cerebrofacial venous metameric syndrome
Cerebrofacial venous metameric syndrome (CVMS) mirrors CAMS with slow-flow venous vascular malformations instead of high-flow arterial malformations.67,68 Common slow-flow vascular malformations seen in CVMS include cavernous malformations and developmental venous anomalies of the brain and craniofacial area. 68 CVMS is an overarching term that encompasses multiple syndromes, the most common of which is Sturge–Weber syndrome. 67
Sturge–Weber syndrome, otherwise known as encephalotrigeminal angiomatosis, is a sporadic congenital neurocutaneous disorder that results from a somatic GNAQ gene mutation. 52 It is considered a variant of CVMS 1 and/or 2. Patients present at birth with a facial CM (port wine stain) typically in the ophthalmic (V1) and/or maxillary (V2) division of the trigeminal nerve and more rarely involving the mandibular (V3) distribution. Retinal capillary VMs can cause glaucoma. Intracranially, leptomeningeal capillary VMs of the ipsilateral cerebral hemisphere lead to chronic venous ischemia of the underlying brain with resulting parenchymal atrophy and cortical and subcortical calcifications. Bilateral Sturge–Weber port wine stains are less common than unilateral; a higher risk of Sturge–Weber syndrome is associated with both bilateral and ophthalmic (V1) division port-wine stains. 69
Capillary malformation–arteriovenous malformation
CM–AVM syndrome is a phakomatosis secondary to familial mutations in the RASA1 gene that result in multiple cutaneous CMs. Affected individuals have multiple 1–2 cm oval-shaped CMs with a reddish-brown to pink appearance and an outer white halo that are randomly distributed across the body, though commonly located on the face or neck. 70 There is substantial phenotypic variability; about one-third of affected individuals also have high-flow AVMs or AVFs, arising in various tissues including brain, spine, and skin/subcutaneous tissue (Figure 3). 47 Limb overgrowth and LMs can also be present. 70
Blue rubber bleb nevus syndrome
Blue rubber bleb nevus syndrome (BRBNS) is characterized by multiple VMs, most commonly of the skin and gastrointestinal tract (Figure 6a–c). 71 Somatic mutations in the TEK gene, which codes for the receptor protein TIE2 (which is involved in the regulation of angiogenesis), are thought to be causative of BRBNS. 72 Involvement of cervicofacial structures in BRBNS is common and includes the tongue, lip, orbit, eyelid, scalp, and neck. 71 Multiple cutaneous VMs are a hallmark feature of BRBNS although malformations can also involve deeper spaces of the head and neck. Lymphatic components and intracranial venous anomalies are also described in BRBNS.73–76 The cutaneous lesions of BRBNS typically do not require intervention; however, such lesions may prompt further imaging to characterize the extent of abnormalities. 73
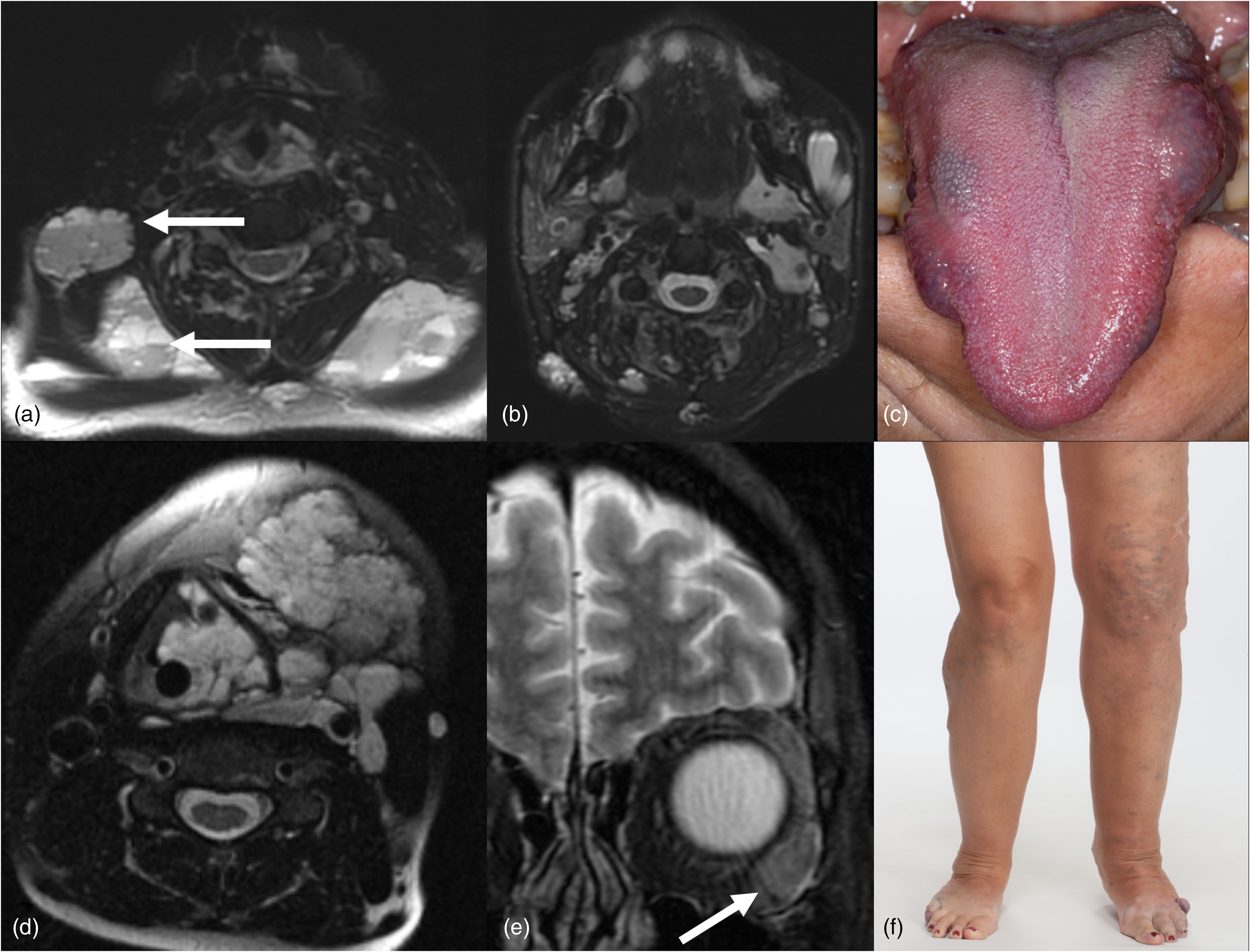
Blue rubber bleb nevus (a–c) and Klippel–Trenaunay syndromes (d–f). Axial T2W STIR images (a and b) demonstrate multiple loculated hyperintense craniofacial soft tissue masses compatible with venolymphatic malformations in a patient with blue rubber bleb nevus syndrome. Lesional fluid–fluid levels are characteristic of lymphatic components (arrows). A clinical photograph shows multiple venous malformations of the tongue (c). Axial T2W STIR image (d) shows a transspatial central and left neck venolymphatic malformation in a patient with Klippel–Trenaunay syndrome. Coronal T2W image (e) shows an additional venous malformation in the lateral left orbit (arrow). Clinical photograph (f) demonstrates varicosities in the left greater than right lower extremities with limb hypertrophy. STIR: short tau inversion recovery; T2W: T2 weighted.
Klippel–Trenaunay syndrome
KTS is a congenital overgrowth disorder that is associated with mutations in the PIK3CA gene (Figure 6d–f). 77 KTS belongs to the PIK3CA-related overgrowth spectrum. 77 Clinically, KTS is characterized by bone and soft tissue hypertrophy of one or more limbs and capillary, venous, and often LMs.2,77,78 Although vascular malformations associated with KTS typically involve the affected limb, additional malformations of the central nervous system have been described, including low-flow vascular malformations such as cavernous malformations. 79
Others
Additional syndromes within the PIK3CA-related overgrowth spectrum that potentially affect the face and neck area are macrocephaly-CM syndrome and CLAPO syndrome with the latter specifically involving CM of the lower lip and LMs in the face and neck. 1 Microcephaly-CM syndrome, a diagnosis confirmed by biallelic pathogenic variants in STAMBP is accompanied by generalized cutaneous CMs, including in the cervicofacial area. 80 Parkes Weber syndrome is also considered a syndrome exhibiting low-flow malformations due to the capillary involvement.
Conclusions
A complex array of craniofacial vascular anomalies exists, each with its own set of clinical and management issues. It is important for the radiologist to be familiar with these entities, recognize the differences between the various vascular malformations, and be familiar with syndromes commonly associated with vascular malformations to optimally contribute to the multidisciplinary care of these children.
Footnotes
Authorship statement
All authors are able to legitimately claim authorship in that all authors met the criteria for authorship as described in the submission guidelines.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed consent
All patients included in this manuscript provided written informed consent for publication.