
Abstract
Background
Informed consent procedures in cluster randomized trials (CRTs) are considerably more complicated than in individually randomized trials. In a CRT, the units of randomization, intervention, and observation may differ in a single trial; there can be multiple levels of participants (individual and cluster level); consent may be required separately for intervention and data collection; and there may be practical constraints to seeking informed consent, for example, due to cluster-level interventions or the sheer size of clusters.
Purpose
We aimed to document consent practices at individual and cluster levels, assess the adequacy of reporting consent in trial publications, and assess associations with two trial characteristics that may influence consent requirements in CRTs: presence or absence of study interventions and presence or absence of data collection procedures at individual and cluster levels.
Methods
We reviewed a random sample of 300 CRTs published during 2000–2008. We sent survey questionnaires to 285 unique authors of these trials to gather detailed information about consent procedures used in each trial.
Results
In all, 182 authors (64%) responded. Overall, 93% (95% confidence interval (CI): 88.8%−96.6%) indicated that participant consent had been sought for some aspects of the study. Consent was less frequently sought for a study intervention (70% of respondents) than for data collection (88%). More than half of the respondents (52%) indicated that consent had been sought at both cluster and individual levels. There was strong evidence for under-reporting of consent in trial publications: only 63% of all trial publications reported that informed consent had been sought for some aspect of the study. The odds ratios (ORs) summarizing the association of the two trial characteristics with cluster-level participant consent were weak (OR = 1.17, p = 0.70 for presence of cluster-level study intervention and OR = 1.54, p = 0.29 for data collection); on the other hand, the ORs summarizing the associations with individual-level consent were strong (OR = 6.2, p < 0.0001 for presence of individual-level intervention and OR = 14.7, p < 0.0001 for data collection).
Limitations
In all, 36% of authors did not respond to the survey; to the extent that consent practices in their trials were different than in respondents’ trials, our results may be biased.
Conclusions
There is a need for improvements in research practices in CRTs as well as their reporting. There may be a lack of clarity about consent requirements at the cluster level in particular. With the publication of the Ottawa Statement on the Ethical Design and Conduct of Cluster Randomized Trials, researchers and research ethics committees now have access to comprehensive ethics guidelines specific to CRTs.
Background
As a general ethical requirement, all research participants must provide written informed consent [1]. We have previously shown that informed consent is less frequently reported in cluster randomized trials (CRTs) than in conventional randomized trials [2]. This gap may reflect a higher level of complexity, practical challenges in seeking informed consent, or uncertainty about appropriate ethical practices in CRTs – including from whom, how, and when consent should be obtained [3]. In patient randomized trials, informed consent procedures are relatively straightforward as the individual patient is simultaneously the unit of randomization, recipient of the intervention, and unit of observation. In a CRT, however, the unit of randomization may be different from the units of intervention and observation. For example, the units of randomization may be hospitals, interventions may be delivered to health professionals, and outcomes may be observed on patients.
Hutton [4] distinguished between consent for randomization, intervention, and data collection. Consent for randomization is different in a CRT than in an individual randomized trial because the unit of randomization is a cluster. For practical reasons, CRT investigators have traditionally approached one or more guardians or ‘gatekeepers’ to provide consent or permission prior to cluster randomization, although the role and authority of the cluster gatekeeper to do so has not been fully understood [5]. Consent for intervention is also more complicated in CRTs because the intervention may be administered to the cluster as a whole, to health or other professionals associated with each cluster, or to individual cluster members themselves. Regardless of the levels of intervention, data may be collected from participants at the cluster level and/or the individual level, or it may be collected from secondary data sources in an anonymous form. CRTs can therefore have participants at multiple levels whose consent may or may not be required. Further discussion of the specific ethical challenges presented in CRTs is presented elsewhere [3,5–8].
In this article, we describe consent practices in a random sample of CRTs published during 2000–2008. We previously reported on consent practices in this sample based only on what was indicated in the trial publication [2]. Here, we report on the results of a follow-up survey of the trial authors in which we elicited more detailed information. Our specific objectives were to (a) describe the prevalence of gatekeeper permission and participant informed consent practices, (b) assess the adequacy of reporting consent in CRTs, and (c) understand the relationship between seeking cluster- and individual-level participant consent and two trial characteristics that are key determinants of consent requirements, namely, whether study interventions and data collection procedures are targeted at those cluster members.
Methods
Identification of sample of trials
Our methodology for identifying the sample of CRTs has been described elsewhere [2]. Briefly, we implemented a highly sensitive electronic search strategy to identify CRTs in MEDLINE [9]. From the results of the search, we selected a random sample of 300 CRTs. A publication was included if it was published in an English language journal between the years 2000 and 2008, and it was the main report of a CRT. Each publication was reviewed by two reviewers who independently abstracted information about trial characteristics and consent procedures, and then reached a consensus.
Design of survey questionnaire
We designed and implemented a web-based survey to gather more detailed information about consent procedures in the sample of trials. The questionnaire consisted of 27 closed-ended questions. We provided open text boxes throughout allowing respondents to elaborate on or explain their answers. In addition to informed consent procedures, the questionnaire gathered information about their experiences with the research ethics review process (reported elsewhere [10]).
We asked authors to identify whether ‘one or more persons at the head of or in charge of each of the clusters’ (i.e., gatekeepers) or anyone else provided ‘agreement or consent’ to the cluster’s involvement in the trial, and to specify who these individuals were. Two separate sections then addressed consent procedures from cluster-level and individual-level participants. The web survey was programmed to skip questions about cluster-level participants if there were no cluster-level participants identified from the publication. We asked whether consent had been sought for any aspect of the study, and when (before or after randomization). If consent had been sought, we asked whether it was for receiving a study intervention and/or for providing data; if consent had not been sought, we asked for an explanation. Authors were asked to indicate whether consent was ‘explicit written’, ‘explicit verbal’, or some ‘other form of consent’. We also asked whether cluster- and individual-level participants in each of the intervention and control arms had been informed about the use of the alternate study arms.
Survey implementation
The methodology for implementing the survey has been described in more detail elsewhere [10]. Briefly, after extensive pilot testing, the survey was administered to 285 unique corresponding authors of the sample of CRTs; for authors with multiple publications in the sample, we used a random number generator to select one publication at random. A series of six contacts was used: a pre-notification email, the survey invitation email, three thank you or reminder emails, and a final postal reminder. The survey was conducted between May and September 2010.
Ethics approval
Authors were informed that participation is voluntary and assured of the confidentiality of their responses. Submission of the questionnaire was considered as consent. The study procedures were approved by the Ottawa Hospital Research Ethics Board.
Data analysis
Data from the survey were merged with the data extracted from the published article. Categorical variables were described using frequencies and percentages; continuous and ordinal variables using medians and inter-quartile ranges. Author, publication, and trial characteristics were compared between survey respondents and non-respondents using chi-squared tests or Wilcoxon two-sample tests, as appropriate.
Following Eldridge et al. [7], we classified each trial into one of four categories depending on the level at which the interventions were delivered: ‘cluster–cluster’ (e.g., community-wide health promotion campaign), ‘external–cluster’ (e.g., intervention involved additional staff such as specialist nurses), ‘professional–cluster’ (e.g., intervention involved training of health professionals), or ‘individual–cluster’ (e.g., intervention was a patient decision aid in breast cancer surgery). In trials that involved cluster–cluster as well as other components, we classified the trial as cluster–cluster. We compared gatekeeper agreement or consent between these categories of trials. Exact or asymptotic methods were used to calculate 95% confidence intervals (CIs) of percentages. Consent procedures reported in the survey were compared to those reported in the published article to assess the prevalence of under-reporting consent. We conducted chi-squared tests to determine the association between seeking of participant consent and two trial characteristics, namely, whether any study interventions or data collection procedures were targeted at the individual or cluster level, as reported in the published article.
Responses in open text boxes were read in combination with responses to closed-ended items. Where possible, we conducted a content analysis by manually assigning codes and tabulating frequencies of a particular response [11] (e.g., number of times a particular reason for not seeking consent was cited). Verbatim comments were selected for illustration. To protect the confidentiality of respondents, information in verbatim comments that could potentially be linked to a trial publication was removed.
Results
Study characteristics
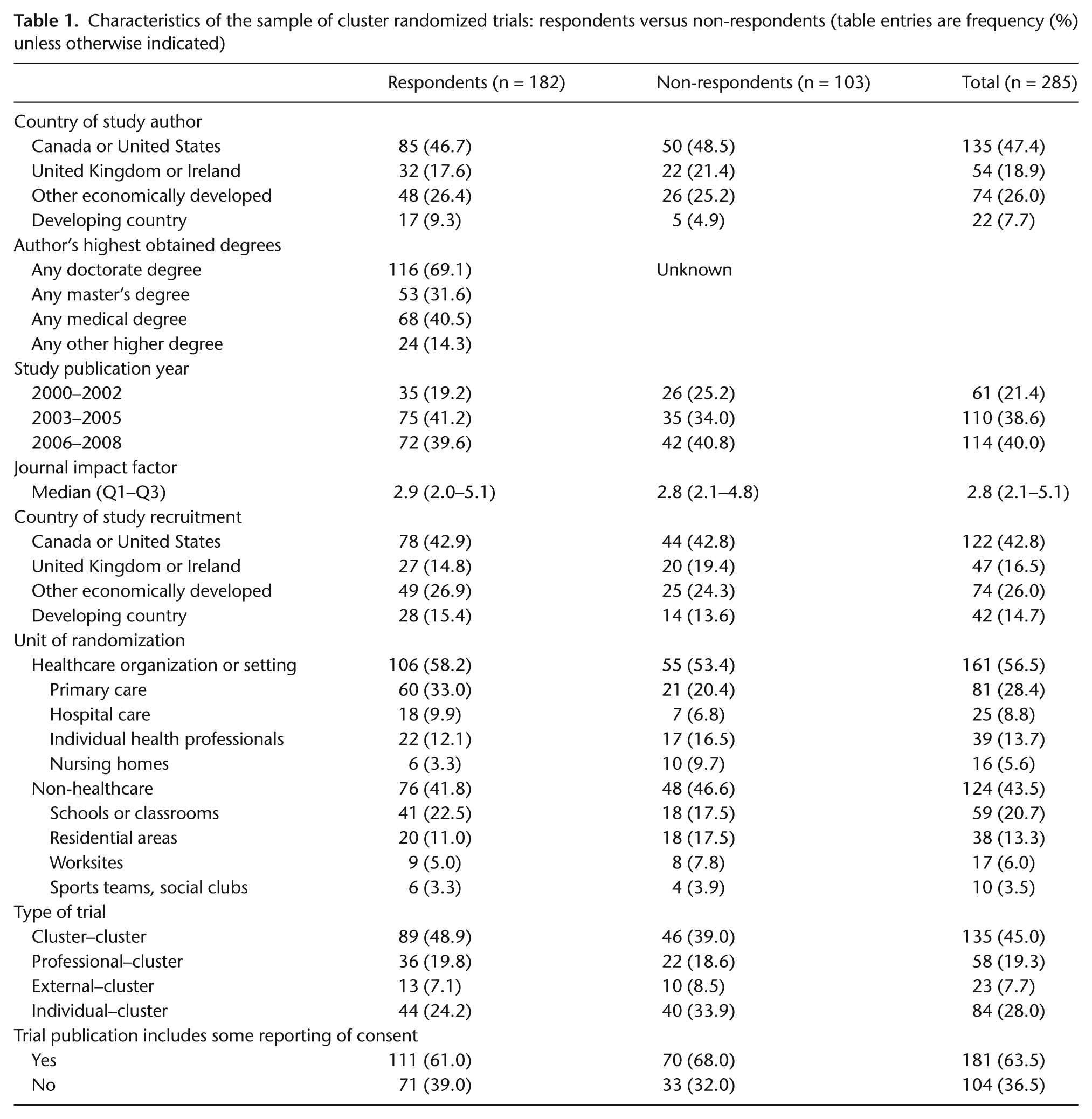
Of the 285 sampled authors, 182 (64%) responded to the survey. Table 1 presents author and study characteristics for respondents and non-respondents. No important differences were observed between the two groups. Most notably, participation in the survey was not associated with reporting of consent in the trial publication: among respondents, 61% of trial publications indicated that some form of consent had been obtained compared to 68% among non-respondents (p = 0.24). Nearly, half of authors resided in Canada or the United States. The unit of randomization was a healthcare organization or setting in close to 60% of trials; nearly one-quarter of trials were in school-based settings. Just over half of trials were cluster–cluster trials; about one-quarter were individual–cluster trials.
Characteristics of the sample of cluster randomized trials: respondents versus non-respondents (table entries are frequency (%) unless otherwise indicated)
Gatekeeper agreement or consent
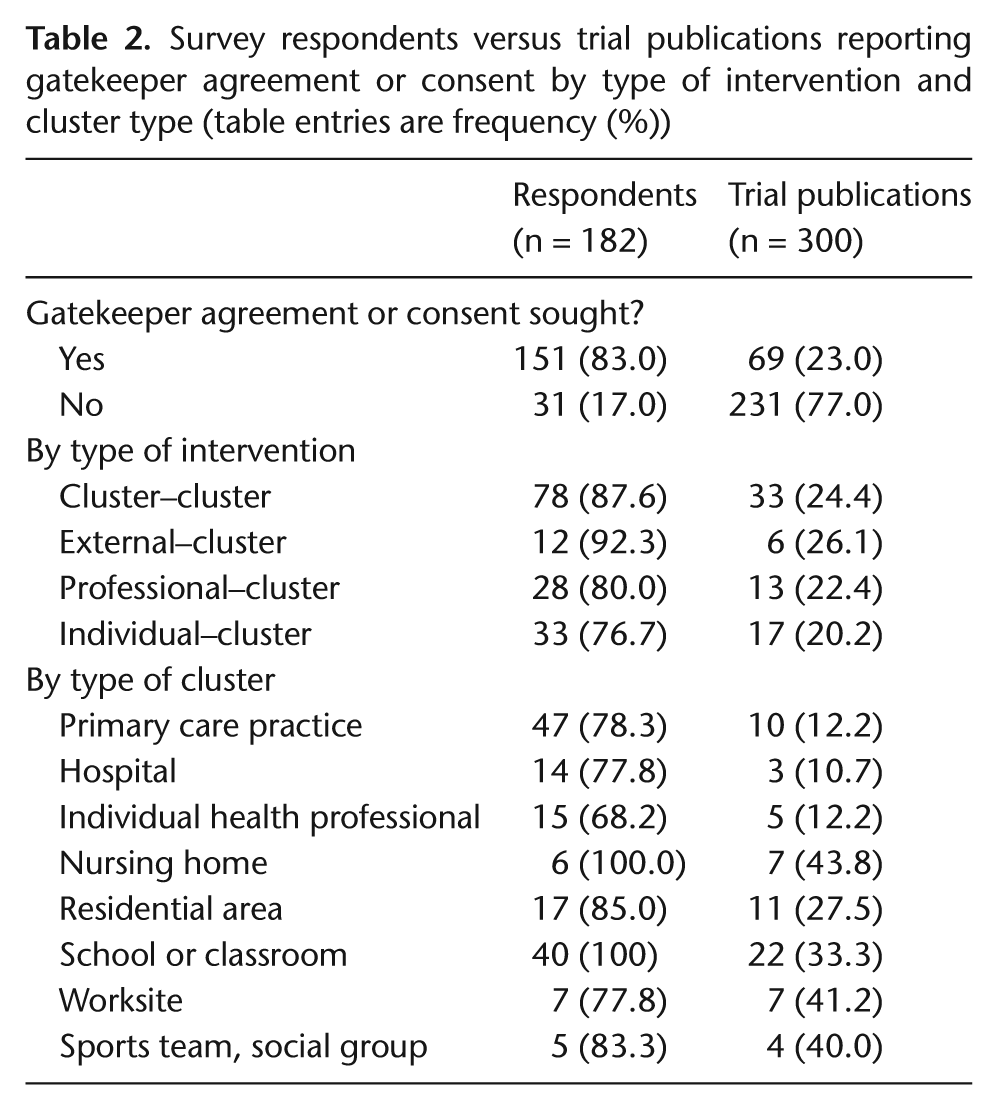
Table 2 presents the prevalence of gatekeeper agreement or consent reported in the survey compared to all trial publications, tabulated by type of intervention and type of cluster. Gatekeepers were common with 151 respondents (83%) indicating use of gatekeepers (although only 23% of all trial publications reported on gatekeepers). The high prevalence of gatekeepers likely reflects the fact that gatekeeper permission is required in order to ‘deliver’ a cluster [5]. When considering gatekeeper prevalence by type of intervention, it was lowest in individual–cluster trials (77%) and highest in external–cluster trials (92%) – not surprisingly as permission from a decision-maker would likely be required to introduce changes in procedures for healthcare delivery. Gatekeeper prevalence varied according to the type of clusters, but they were least common when individual health professionals were the units of randomization (68%). In such trials, health professionals themselves may have been participants and may have been required to provide informed consent, perhaps lessening the need for gatekeeper permission.
Survey respondents versus trial publications reporting gatekeeper agreement or consent by type of intervention and cluster type (table entries are frequency (%))
Although we did not request details about the role of the cluster gatekeeper, respondents indicated in open text boxes that the identified gatekeepers had provided ‘agreement on behalf of the cluster’, ‘consent to the cluster’s involvement’, ‘consent on behalf of trial participants’, ‘formal approval to conduct the study’, ‘endorsement of the study’, ‘study advice’, ‘permission to approach participants’, ‘support and oversight’, ‘approval of use of personal data’, ‘facilitation of discussion with community members’, ‘liaison with a community advisory board’, and ‘approval of the study design’. The nature of gatekeeper permission ranged from written approval, contracts and legal agreements, to informal verbal consent.
Details of participant consent: survey questionnaire versus trial publication
Of the 285 trials included in the survey, we could identify potential cluster-level participants in 221; the questionnaires sent to these authors therefore included questions pertaining to cluster-level consent procedures. A total of 182 respondents answered questions relating to individual-level consent procedures; of these, 147 received the additional questions relating to cluster-level consent procedures.
Table 3 presents detailed information about participant consent practices in the sample of trials, including whether or not consent had been obtained for any aspect of the study, for study interventions or for data collection specifically, and whether it had been obtained from individual-level or cluster-level participants or both. Where possible, proportions are presented for survey respondents versus all trial publications. To simplify the analyses and facilitate coding of composite variables, three respondents with missing information were classified as not having sought consent. This was considered a reasonable assumption as trialists would have likely indicated consent if indeed it had been sought. Overall, 170 respondents (93%, 95% CI: 88.8%−96.6%) indicated form of participant consent for some aspects of the study, compared to only 63% (95% CI: 57.9%−68.8%) of trial publications reporting participant consent. Consent was less common for a study intervention (70% of respondents) than for data collection (88% of respondents). More than half of the respondents (52%) indicated that consent had been sought at multiple levels. Comparable proportions for the full set of trials, that is, with respect to what consent was for and at what levels, could not be presented because few study publications provided these details.
Survey respondents versus trial publications indicating consent from individual- and/or cluster-level participants (table entries are frequency (%))
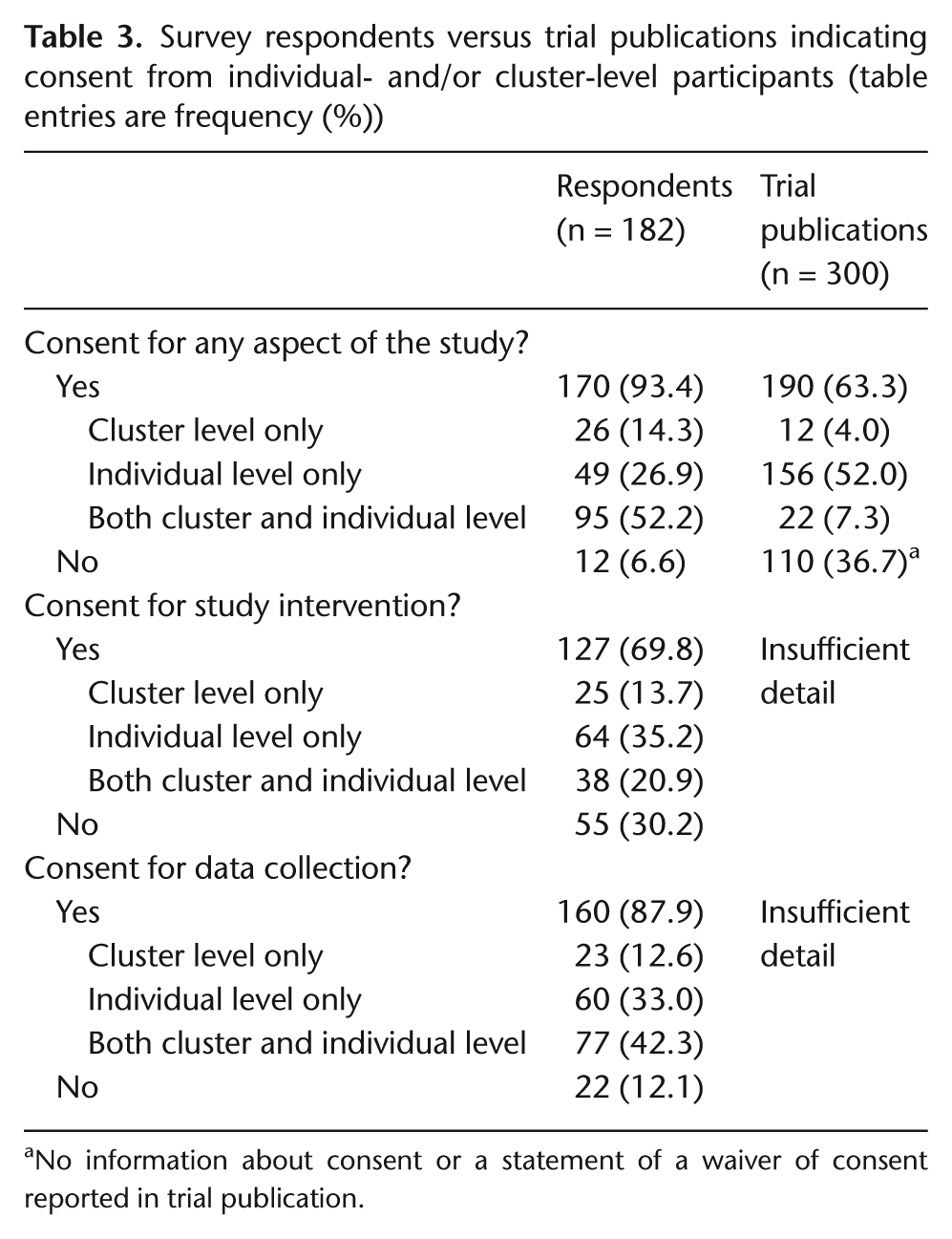
No information about consent or a statement of a waiver of consent reported in trial publication.
Timing of individual- and cluster-level consent
Table 4 presents the prevalence and timing of cluster-level and individual-level participant consent in the intervention and control arms of the study. Among those trials with cluster-level participant consent, consent was more frequently sought before randomization than after randomization, but among those with individual-level participant consent, the opposite was true: consent was more frequently sought after randomization than before. With respect to the intervention arm in particular, 71% of trials with cluster-level consent obtained this consent before randomization, but only 25% of trials with individual-level consent obtained this consent before randomization. This likely reflects practical difficulties in identifying individual cluster members prior to cluster randomization. Surprisingly, consent prevalence was only slightly higher in the intervention than in the control arm.
Prevalence and timing of cluster- and individual-level participant consent procedures in respondent cluster randomized trials: intervention and control arms
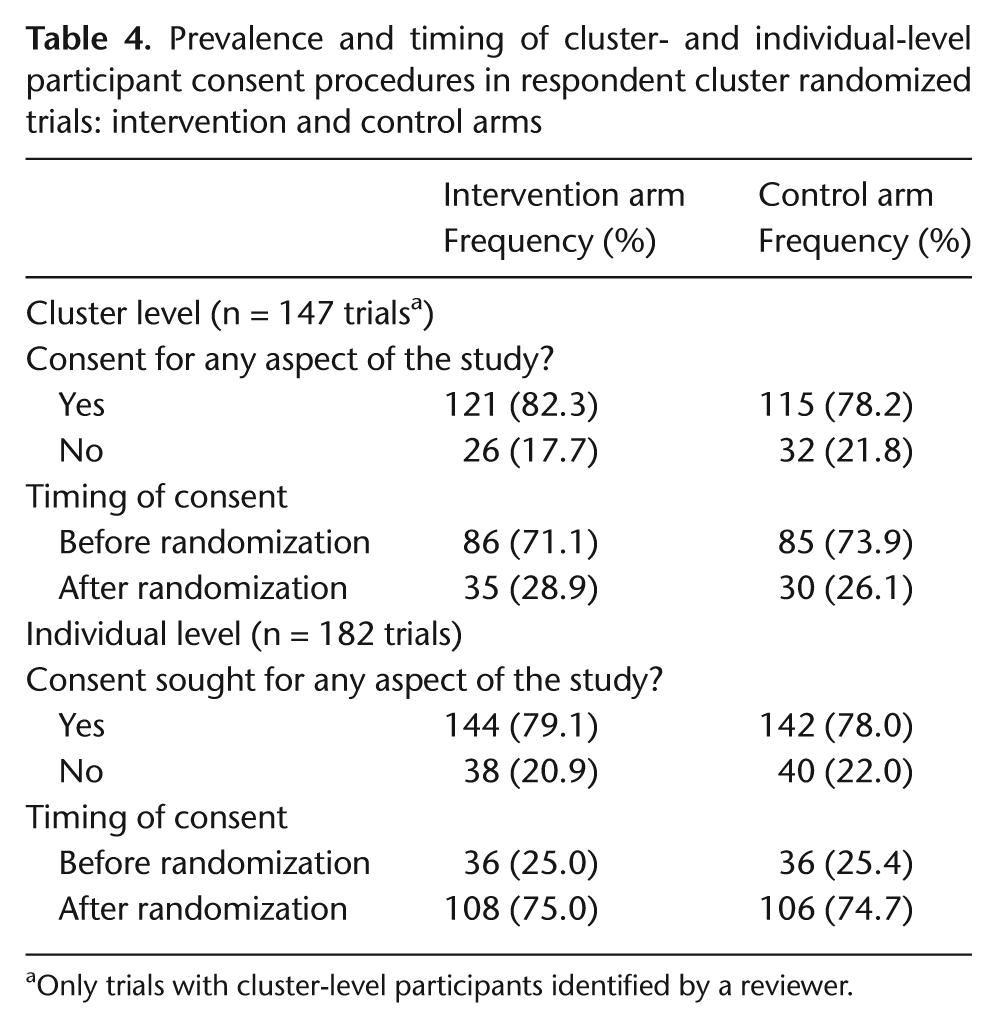
Only trials with cluster-level participants identified by a reviewer.
Reasons for not seeking consent
Of the 26 respondents who indicated that they had not sought consent from cluster-level participants in the intervention arm, 18 offered an explanation, including that participants had been given the opportunity to opt-out (7); they were not considered research subjects (3) or were not targeted by study procedures (2); the intervention was an educational program (3), quality improvement intervention (2), or system-level change (2) and participation was required or expected based on professional duties (1); procedures met ethical requirements (2); data collection was anonymous (1); and gatekeepers had given permission on behalf of participants (5). Note that some respondents indicated more than one reason. Selected verbatim explanations are presented in Table 5.
Selected respondent explanations for why consent had not been sought
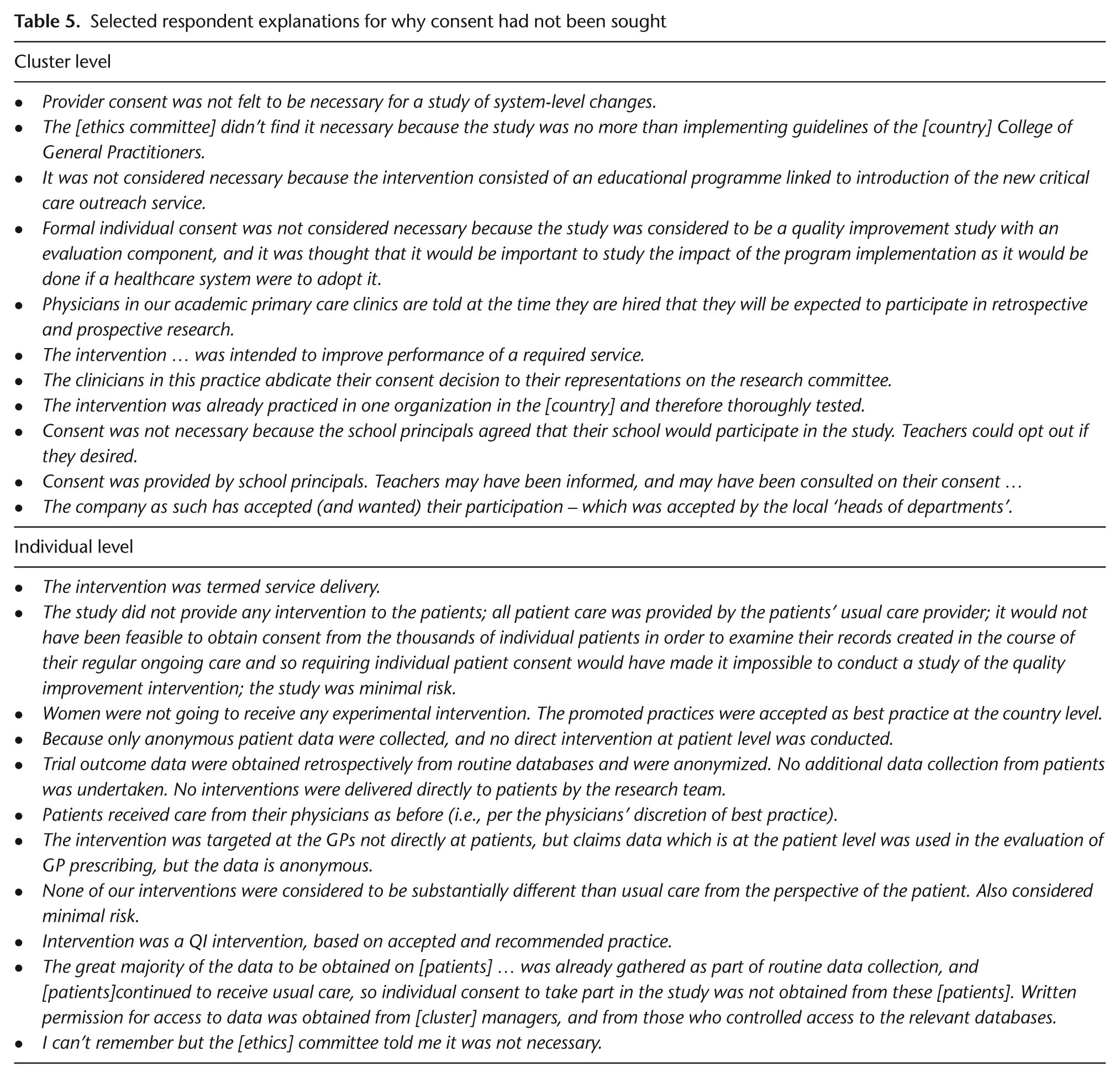
Of the 38 survey respondents who indicated that they had not sought consent from individual-level participants in the intervention arm, 28 offered an explanation, including that study procedures were not directly targeting these participants (13 respondents) or there was minimal involvement from participants (2); interventions were usual procedures or standards of care (7), best practices (2), or quality improvement interventions (3); there was minimal risk to participants (1) or they were given the opportunity to opt out (4); consent was not practically possible (2); data collection was from secondary sources (4) or anonymous (5); and procedures met ethical requirements (5) or waivers had been obtained (3). Selected verbatim explanations are presented in Table 5.
Types of consent
Table 6 presents prevalence of written, verbal, or other consent procedures from cluster- and individual-level participants. Among the 147 respondents answering questions about cluster-level participants, 83 (57%) indicated some form of cluster-level consent for a study intervention; however, only 43 (29%) indicated that this was ‘explicit written’ consent. In all, 100 (68%) indicated cluster-level consent for data collection, but only 60 (41%) indicated that this was ‘explicit written’ consent. Among all 182 respondents answering questions about individual-level participant consent, 102 (56%) indicated individual-level consent for a study intervention; 87 (48%) ‘explicit written’ consent. A total of 137 (75%) indicated some form of individual consent for data collection, with 110 (60%) indicating ‘explicit written’ consent. Explanations provided in open text boxes for ‘other’ consent, included secure e-mail communication, a contact person had been asked to secure the consent of individuals, return of survey questionnaires or participation in a telephone survey was regarded as consent, consent was covered by an annual consent or permission form, and it was passive consent or assent from students.
Type of participant consent procedures in respondent cluster randomized trials
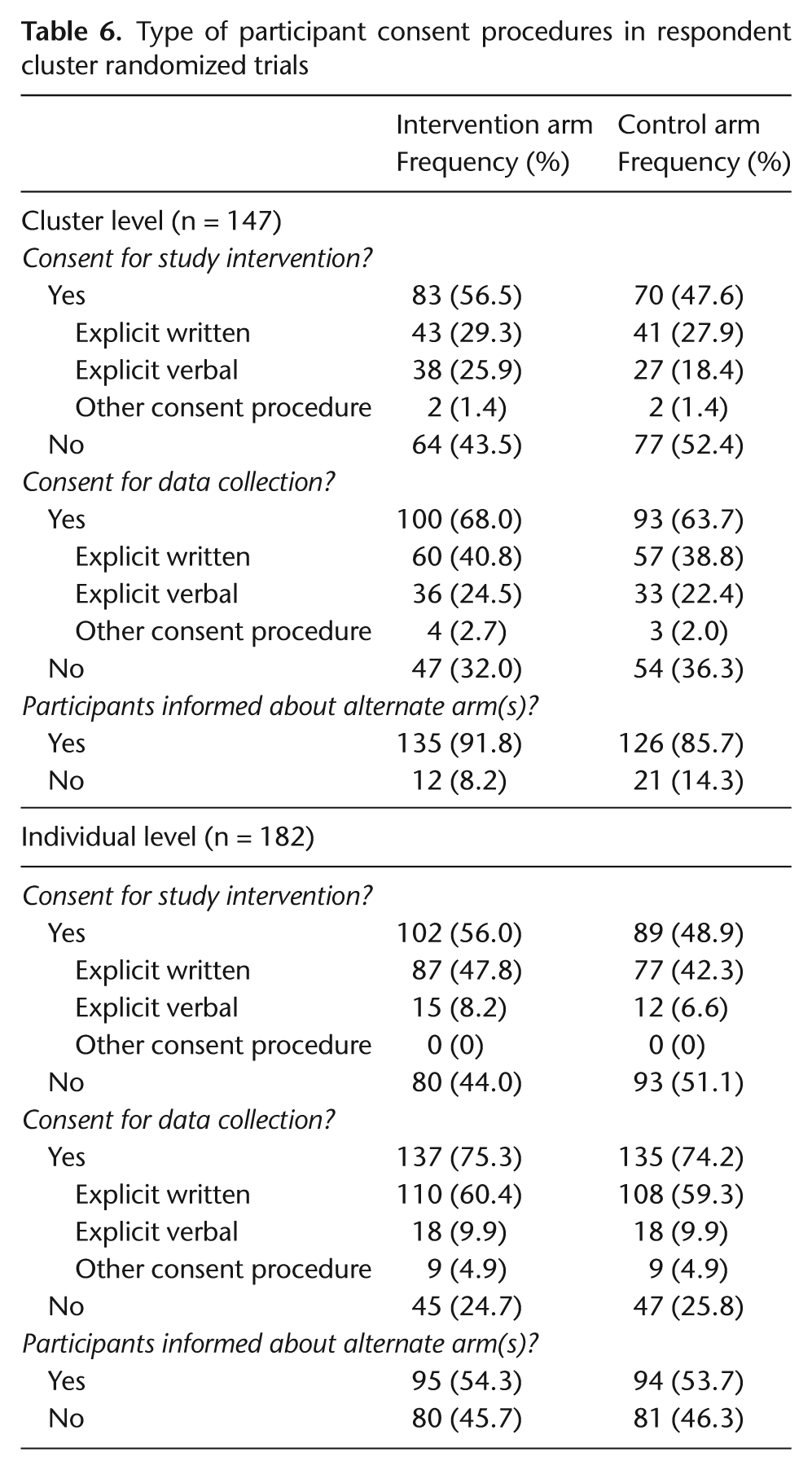
Cluster-level participants in the intervention arm had been informed about the alternate arm in 135 (92%) of respondents’ trials; in the control arm, the corresponding number was 126 (86%). Proportions of trials with individual-level participants informed about the alternate arm were lower (54% in both arms).
Associations with trial characteristics
Table 7 presents the relationship between cluster- and individual-level consent reported in the survey and two key characteristics of CRTs extracted from the trial publication. Among trials with experimental interventions targeting the cluster-level participants, 67 (57%) had sought some form of consent from cluster-level participants for the intervention; among those with data collection procedures at the cluster level, 32 (74%) had sought consent from cluster-level participants for data collection. The odds ratios (ORs) summarizing the associations of these trial characteristics with cluster- level consent were weak (OR = 1.17, p = 0.70 for intervention and OR = 1.54, p = 0.29 for data collection). This may reflect lack of clarity about consent requirements at the cluster level.
Association between survey-reported participant consent procedures and publication details of study interventions and data collection procedures (table entries are frequency (%) unless otherwise indicated)
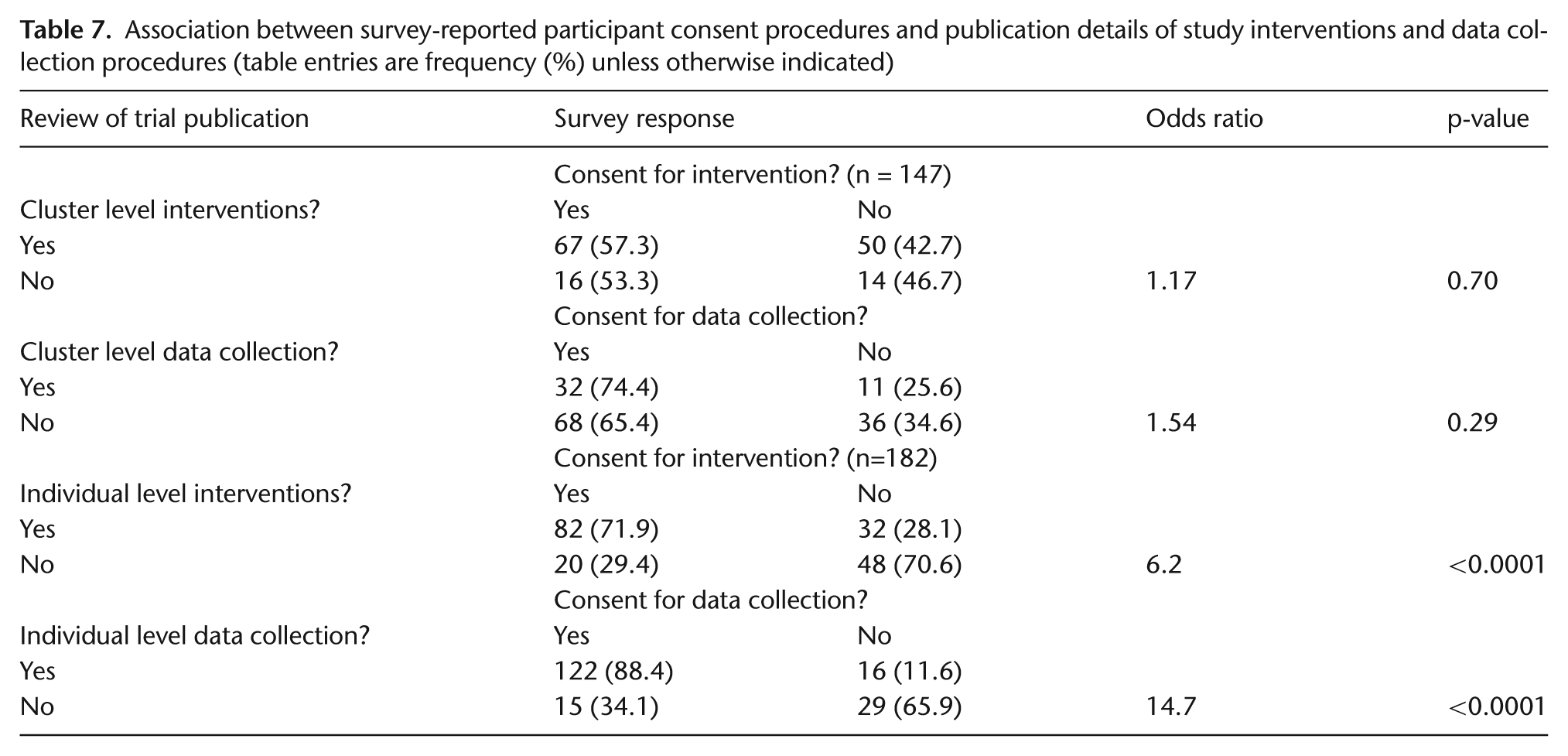
Among trial publications with experimental interventions targeting the individual-level participants, 82 (72%) had sought some form of individual-level consent for the intervention; among those with data collection procedures at the individual level, 122 (88%) had sought individual-level consent for data collection. The ORs summarizing the associations of these trial characteristics with individual-level consent were strong (OR = 6.2, p < 0.0001 for intervention and OR = 14.7, p < 0.0001 for data collection).
Discussion
Until the recent publication of the Ottawa Statement on the Ethical Design and Conduct of Cluster Randomized Trials [12,13], investigators lacked detailed guidance to determine from whom, when, and how informed consent ought to be obtained in CRTs. In our survey of 182 authors of published CRTs (conducted prior to the publication of the Ottawa Statement), we found that no participant consent had been obtained for study interventions in 30% of the trials, and no participant consent for data collection in 12%; in 7% of trials, no participant consent had been obtained for any aspect of the trial. Although we did not undertake to make a judgment as to the appropriateness of consent practices in each trial, the Ottawa Statement generally requires all research participants, whether at the individual or the cluster level, to provide informed consent. A research participant is defined as any cluster member who is the recipient or the direct target of a study intervention (whether an individual- or cluster-level intervention), with whom researchers interact for study purposes, or about whom identifiable private information is collected [14,15]. Based on the results from our survey, nearly 1 in 2 trials with experimental interventions targeting cluster-level participants had no consent for the intervention from cluster-level participants, and 1 in 4 with data collection procedures at the cluster level had no consent for data collection at that level. In addition, 1 in 4 with experimental interventions targeting individuals had no consent for the intervention from individual-level participants, and 1 in 8 with data collection interventions at the individual level had no consent for data collection at that level. These rates of consent failures would be even higher if considering only written forms of informed consent. According to the Declaration of Helsinki, research subjects must provide freely given informed consent, preferably in writing [1]. If the consent cannot be expressed in writing, the non-written consent must be formally documented and witnessed.
It is possible that there were legitimate reasons for not seeking consent or practical constraints preventing consent (e.g., large cluster sizes). According to the Ottawa Statement, waivers or alterations of consent (alteration or deletion of some of the standard elements of disclosure) can be justified only when the study would not otherwise be feasible and when the risks involved are minimal [12]. Feasibility will depend on a variety of factors, including cluster size and other logistical limitations. However, only 16 study publications (5%) indicated that waivers of consent for any aspect of the study had been obtained, and few respondents mentioned waivers or minimal risk in their explanations for why consent had not been sought. An alternative explanation is that researchers and ethics committees may have been satisfied with gatekeepers providing proxy consent on behalf of participants. However, the Ottawa Statement does not permit gatekeepers to provide proxy consent on behalf of individuals in a cluster.
Consistent with our findings, a similar survey of authors of 113 CRTs [16], found that 12% did not seek participant informed consent. Further comparison between these two surveys is not possible due to differences in the data collection instruments: for example, our questionnaires separately addressed procedures for individual- and cluster-level participants, whereas the earlier survey did not specifically identify participants at the individual and cluster levels. Yet, more than half of respondents in our survey indicated that they had sought consent from multiple levels.
Unlike Eldridge et al. [7], we found strong evidence for under-reporting in trial publications: under-reporting was common for gatekeeper permission (83% vs. 23%) as well as for trial participant consent (93% vs. 63%). In addition, important details with respect to from whom, when, and for what consent had been obtained were lacking in the publication.
Our study has several limitations. First, as in most surveys, it is subject to the risk of non-response bias. Our response rate was 64% (similar to the 65% response rate in the earlier survey) [16]. We investigated differences between respondents and non-respondents, but no important differences were observed. Second, we only questioned trialists about cluster-level participant consent procedures if our review of the trial publication indicated the presence of such participants. This was done by a single reviewer, and it is possible that we may have misidentified the presence of cluster-level participants. However, only two respondents indicated that the individuals we had identified were in fact not participants in the trial. Third, most CRTs are complex, and it is possible that data abstractors misclassified characteristics of the trial publications. We tried to minimize this risk by using two independent reviewers. Fourth, our estimates may be subject to other forms of bias, such as recall bias: authors may not have been able to recall detailed procedures in trials conducted as many as 10 years ago. Authors who indicated that they could not recall or who had a missing response, were classified as not having sought consent; this may have caused a further downward bias in our estimates of consent prevalence. On the other hand, an upward ‘social desirability’ bias is possible if authors did not want to be found to be negligent in this important ethical requirement.
Results from our survey confirm that reporting of consent in CRTs is inadequate [2]. In addition, our survey shows that improvements are required in the ethical conduct of CRTs, particularly with respect to cluster-level consent procedures. We previously reported on difficulties in the research ethics review process of these trials from the perspectives of the study authors [10]. With the publication of the Ottawa Statement, researchers and research ethics committees now have explicit guidelines to aid the design, conduct, and review of CRTs. It is our hope that the Ottawa Statement will lead to improvements, both in the research ethics review process, and in the ethical conduct of CRTs.
Footnotes
Acknowledgements
The web survey was designed by senior programmer Dong Vo at the Methods Centre of the Ottawa Hospital Research Institute (OHRI). We thank the authors of the sample of trials for taking the time to complete our survey questionnaire.
Funding
This work was supported by the Canadian Institutes of Health Research (grant number MOP85066, MOP89790).
Conflict of interest
None declared.