
Abstract
Introduction:
Participant noncompliance, in which participants do not follow their assigned treatment protocol, has long complicated the interpretation of randomized clinical trials. No gold standard has been identified for detecting noncompliance, but in some trials participants’ biomarkers can provide objective information that suggests exposure to non-study treatments. However, existing methods are limited to retrospectively detecting noncompliance at a single time point based on a single biomarker measurement. We propose a novel method that can leverage participants’ full biomarker history to detect noncompliance across multiple time points. Conditional on longitudinal biomarker data, our method can estimate the probability of compliance at (1) a single time point of the trial, (2) all time points, and (3) a future time point.
Methods:
Across time points, we model the biomarker as a mixture density with (latent) components corresponding to longitudinal patterns of compliance. To estimate the mixture density, we fit mixed effects models for both compliance and the biomarker. We use the mixture density to derive compliance probabilities that condition on the longitudinal biomarker data. We evaluate our compliance probabilities by simulation and apply them to a trial in which current smokers were asked to only smoke low nicotine study cigarettes (Center for the Evaluation of Nicotine in Cigarettes Project 1 Study 2). In the simulation, we investigated three different effects of compliance on the biomarker, as well as the effect of misspecification of the covariance structures. We compared probability estimators (1) and (2) to those that ignore the longitudinal correlation in the data according to area under the receiver operating characteristic curve. We evaluated estimator (3) by plotting its calibration lines. For Center for the Evaluation of Nicotine in Cigarettes Project 1 Study 2, we compared estimators (1) and (3) to a probability estimator of compliance at the last time point that ignores the longitudinal correlation.
Results:
In the simulation, for both compliance at the last time point and at all time points, conditioning on the longitudinal biomarker data uniformly raised area under the receiver operating characteristic curve across all three compliance effect scenarios. The gains in area under the receiver operating characteristic curve were smaller under misspecification. The calibration lines for the prediction of compliance closely followed 45°, though with additional variability under misspecification. For compliance at the last time point of Center for the Evaluation of Nicotine in Cigarettes Project 1 Study 2, conditioning on participants’ full biomarker history boosted area under the receiver operating characteristic curve by three percentage points. The prediction probabilities somewhat accurately approximated the non-longitudinal compliance probabilities.
Discussion:
Compared to existing methods that only use a single biomarker measurement, our method can account for the longitudinal correlation in the biomarker and compliance to more accurately identify noncompliant participants. Our method can also use participants’ biomarker history to predict compliance at a future time point.
Keywords
Introduction
Participant noncompliance, in which participants do not follow their randomly assigned treatment protocol, has long complicated the interpretation and conduct of randomized clinical trials. Participant noncompliance often occurs when participants must self-administer the treatment without the supervision of study personnel. 1 Given this autonomy, participants may deviate from their treatment protocol for numerous reasons, including advent of side effects, insufficient benefit, and availability of commercial alternatives to the treatment. 2 Examples of trials with participant noncompliance include regulatory tobacco trials of very low nicotine content cigarettes, where current smokers must only smoke the study cigarettes; 3 and opioid dependence trials, where patients with substance abuse disorders must self-inject study depots.4,5 Furthermore, in longitudinal studies, multiple time points present multiple opportunities for participants to be noncompliant.
Compared to compliant participants, noncompliant participants do not receive the full dose of treatment and as such may have poor study outcomes. This may dilute the intention-to-treat (ITT) estimate. 6 In addition, self-reported noncompliance may systematically differ from actual noncompliance according to some confounding variable. 7 This may create a difference between the Per Protocol estimate based on self-reported compliance and the treatment effect if all participants had complied (i.e. the causal effect). 8 As a motivating example, in the United States, the Family Smoking Prevention and Tobacco Control Act provides the Food and Drug Administration with the regulatory authority to reduce (but not eliminate) the nicotine content in commercial cigarettes if it would improve public health. Regulatory tobacco trials seek to evaluate the effect of such potential changes to commercial cigarettes. As these trials investigate interventions that could be mandated by federal law to force compliance, the causal effect is more relevant than the ITT estimator. 9
Modeling longitudinal compliance is thus important for the following reasons. First, for completed trials, identifying compliant participants enables estimation of the causal effect, 10 which may be different than both the ITT and Per Protocol estimates. Methods are thus needed to identify compliant participants to properly weigh the study outcomes and adjust for confounding, but only those for a single time point have been proposed.11,12 Second, noncompliant participants are more likely to drop out. 6 Methods are thus needed to identify noncompliant participants during the trial for remedial intervention.
However, in many therapeutic areas, no gold standard has been identified for detecting participant noncompliance. Trial designers must frequently rely on imperfect measures of participant noncompliance based on subjective or indirect information (e.g. self-reported compliance).13,14 Moreover, detecting participant noncompliance falls into the broader category of diagnosis without a gold standard, a subject of considerable study. With respect to true (but unobserved) disease status, statistical methods have been able to both estimate summary measures (e.g. disease prevalence) and individually model disease progression.15,16 Yet, few statistical methods exist for diagnosing patients whose disease status (in our case, compliance) may shift back and forth between two disease states.
In some trials, participants’ biomarkers may systematically change in response to the treatment or any alternatives to provide objective information about noncompliance. Consider three recently published regulatory tobacco trials studying the effect of very low nicotine content cigarettes (with 0.4 mg of nicotine per gram of tobacco) on smoking behavior.3,17,18 In these trials, current smokers were randomized to smoke either normal nicotine content cigarettes (with 15.8 mg of nicotine per gram of tobacco) or very low nicotine content cigarettes provided by the study for 6 or 20 weeks. During the follow-up period, participants were asked to only smoke their assigned study cigarettes but could additionally smoke commercial cigarettes (i.e. non-study cigarettes) with normal nicotine content. Participants who smoked non-study cigarettes were considered to be noncompliant. Most participants self-reported compliance at each time point.
At each follow-up visit, participants were asked to give samples of various biomarkers including total nicotine equivalents, which measures most nicotine metabolites in the urine to evaluate recent nicotine exposure. Based on the findings of a previous study of participants who were sequestered in a hotel and only had access to very low nicotine content cigarettes, only 5% of participants randomized to very low nicotine cigarettes were expected to have total nicotine equivalents above 6.41 nmol/mL if they were fully compliant. 19 However, in one trial, 63% of participants who self-reported compliance at week 6 had total nicotine equivalents above 6.41 nmol/mL. 20
In randomized trials of very low nicotine content cigarettes, biomarkers like total nicotine equivalents can suggest exposure to non-study cigarettes. 21 In the absence of a gold standard for compliance, Boatman et al. 11 developed a method for estimating the probability of compliance at a single time point based on biomarker data collected at that time point. However, this method cannot leverage biomarker data collected at previous time points, and nor can it predict compliance at a future time point.
We propose a longitudinal method that uses biomarker data collected across multiple time points to estimate longitudinal compliance when the true compliance status is not directly observed. Specifically, we model the biomarker as a mixture density whose (latent) components correspond to different compliance patterns over time, a modeling approach similar to growth mixture models. 22 Our method can estimate the probability of compliance at (1) a single time point of the trial, (2) all time points, and (3) a future time point. We first evaluate our method by simulation, examining two factors: (1) the effect of compliance on the biomarker and (2) incorrect specification of the covariance structures of the models fit. Second, we apply our method to the data collected in the Center for the Evaluation of Nicotine in Cigarettes Project 1 Study 2 (CENIC-P1S2) trial.
Methods
Overview
For our purposes, we treat the true compliance status as unobserved and binary; that is, each participant can either fully comply or not fully comply at each time point. To derive the probability of compliance at a single time point conditional on a single biomarker measurement, Boatman et al. modeled the distribution of the biomarker as a two-component mixture density of compliance and noncompliance and then used Bayes’ rule to derive the desired probability of compliance from the mixture density. 11 Our goal is to generalize this approach to multiple time points to identify if and when participants are noncompliant. In the longitudinal setting, the number of components in the mixture density exponentially increases beyond two as participants can shift between compliance and noncompliance at each time point. In addition, multiple observations on each participant present the possibility of correlated data. In the following section, we explain how to extend the mixture density to multiple time points while allowing for correlation among repeated measurements.
Modeling the mixture density
Let
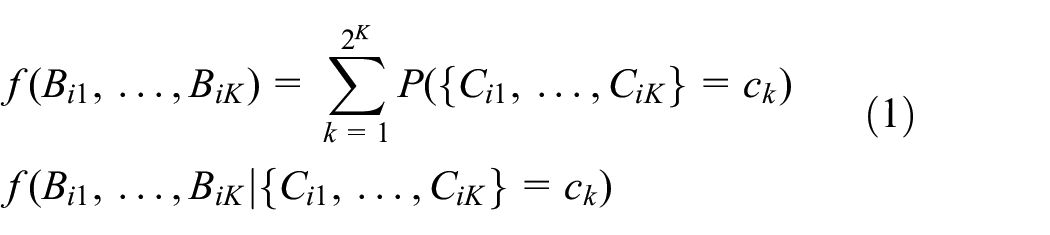
A number of longitudinal models can be fit to
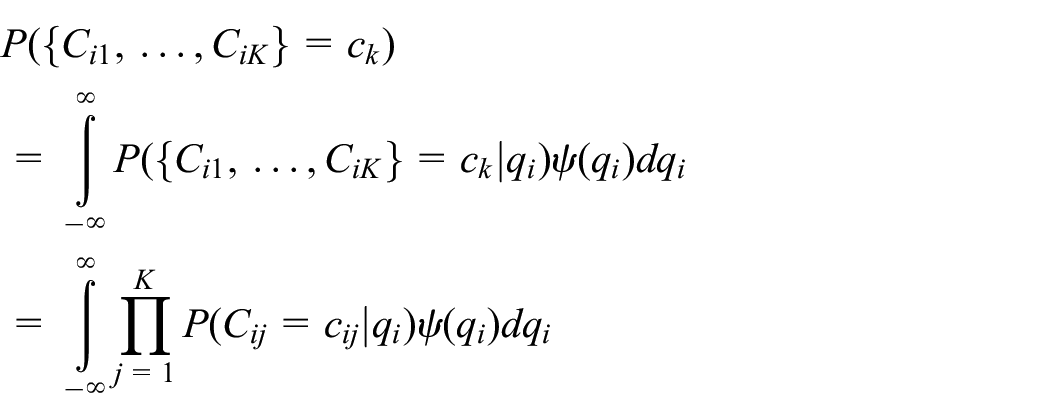
where
Similarly, we can use the conditional independence assumption to write
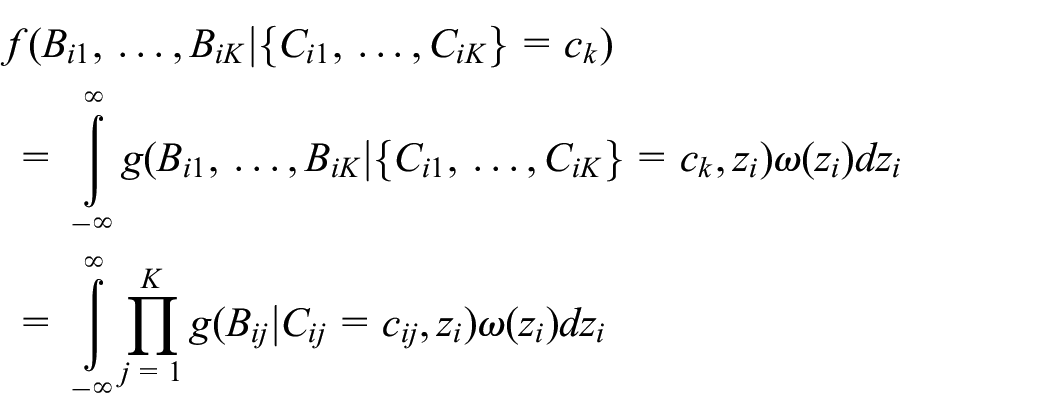
where
Our model setup allows us to select link functions and densities for

where
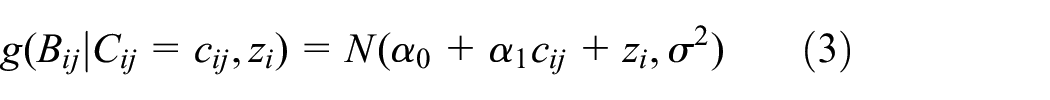
where
Our method depends on properly specifying both the covariates and covariance structures for both the compliance and biomarker mixed effects models. As our method is fully likelihood-based, any number of likelihood-based tests and information criteria can be used to select which models to fit. In the application to CENIC-P1S2, we will use Bayesian information criterion (BIC) to investigate whether or not to include a fixed effect for time in the compliance mixed effects model. 23 While likelihood-based tests and information criteria can help guide model selection, the models fit may not be properly specified. Moreover, previous research of latent models has found that model misspecification of the covariance structures can lead to bias and variability in the parameter estimates.24,25 In the simulation, we will investigate the effect of misspecifying the covariance structures on model fit.
Let
One advantage of this approach is that we can include participants who lack biomarker data at some time points. That is, mixed effects models estimated by maximum likelihood can inherently handle data that are missing at random. Given this assumption, estimators of
Deriving compliance probabilities of interest
We can use the mixture density to derive compliance probabilities that condition on the longitudinal biomarker data. Intuitively, these compliance probabilities should be more accurate than methods which only condition on a single biomarker measurement. Examining all time points, we can use Bayes’ rule to derive the probability of compliance at all time points conditional on the biomarker history as
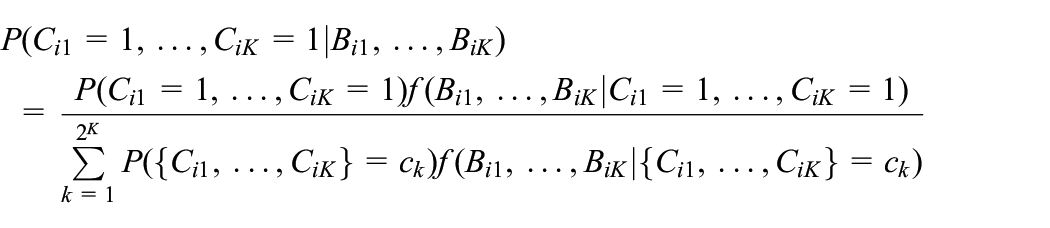
Note that both
In addition, by summing over the relevant posterior compliance probabilities, we can derive the probability of compliance at the last time point conditional on the biomarker history as
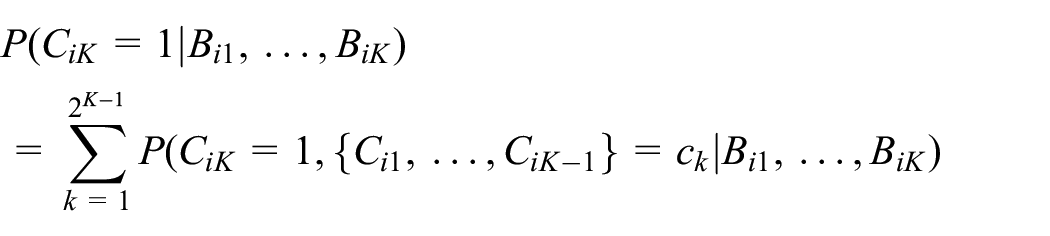
Assuming that we are given biomarker data up to but not including the last time point, we can integrate out the future unobserved biomarker value to derive the prediction probability of compliance at the last time point as
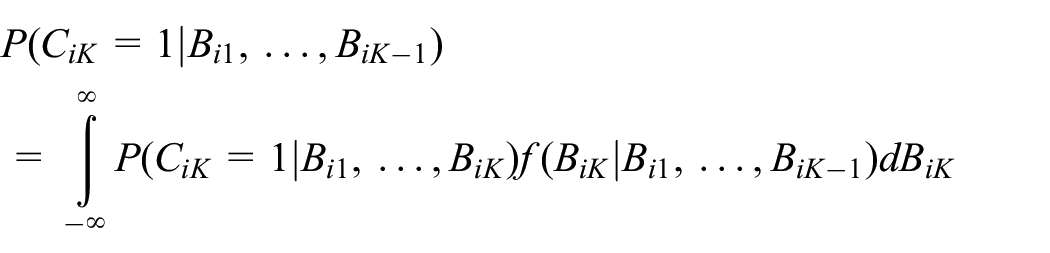
where:
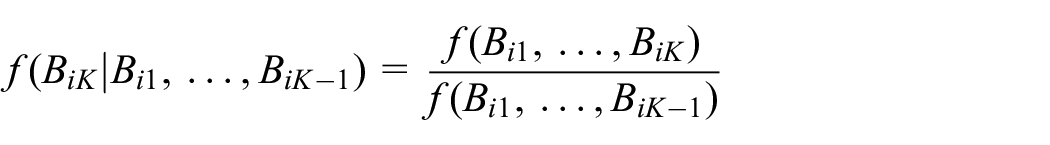
Note that both the numerator and denominator of the above fraction are simply the mixture density in equation (1) given
Simulation study design
We used Monte Carlo simulation to assess the accuracy of our method in discriminating between compliers and noncompliers as well as predicting compliance at a future time point. We generated longitudinal compliance data from the mixed effects probit model outlined in equation (2), where there are no covariates and a single participant-specific random intercept. Specifically, we set
We generated longitudinal biomarker data from the linear mixed effects model outlined in equation (3), where there is an indicator variable for compliance at the given time point and a single participant-specific random intercept. Specifically, we set the intercept of noncompliant participants to be
To set the effect of compliance on the mean biomarker value (i.e.
To mimic the data collected in CENIC-P1S2, we set
In addition, for both simulations of
To evaluate our method, we compared the average parameter estimates to the true values, where only comparisons for
To assess the accuracy of our method in detecting noncompliance, we first compared the AUC values of the various compliance probabilities under the true parameters to the AUC values under the estimated parameters. Moreover, to protect against model overfit, AUC values were estimated from large, fixed independent test sets (i.e. we used Monte Carlo integration) as closed-form solutions are not available.
In addition, to measure the extent to which conditioning on longitudinal biomarker data improves detection of noncompliance, we compared the AUC values of
To assess the accuracy of
Application to CENIC-P1S2
We applied our method to the CENIC-P1S2 trial, which sought to evaluate the effect of very low nicotine content cigarettes with and without a nicotine patch on number of cigarettes smoked per day (NCT02301325). 18 Current smokers who had no intention of quitting were randomized in a 2×2 factorial design, with very low nicotine content versus normal nicotine content cigarettes as the first factor and nicotine patch versus no nicotine patch as the second factor. During the follow-up period of 6 weeks with study visits every week, participants were asked to only smoke the study cigarettes provided by the trial but could additionally smoke commercial cigarettes.
Although previous trials of very low nicotine content cigarettes have used total nicotine equivalents as a biomarker for detecting noncompliance, nicotine patches elevate nicotine levels regardless of compliance to very low nicotine content cigarettes; therefore, we cannot use total nicotine equivalents to identify noncompliant participants in CENIC-P1S2. Instead, we used the tobacco alkaloid anatabine which has a weaker association with compliance than total nicotine equivalents.28,29 Due to the skewness of the distribution of anatabine, and the fact that it is a concentration, we modeled it on the natural logarithm scale. In addition, some participants had observations of anatabine that fell below the limit of quantification and were thus censored. For these participants, we modified the likelihood of the conditional biomarker to accommodate left-censored observations. 30
A total of
In the model fit, we sought to determine whether or not compliance changed across time points. We compared the BIC of the models with a single compliance intercept versus a compliance intercept for each of the six time points. In addition, to better understand the relationship between the longitudinal anatabine values and the compliance probabilities, we constructed a spaghetti plot of log(anatabine) shaded by the probability of compliance at all time points. Similar to the simulation study, we compared
To assess
Results
Simulation study
For the simulation scenarios where
Estimated bias and Monte Carlo standard deviation (MC SD) of parameter estimates for
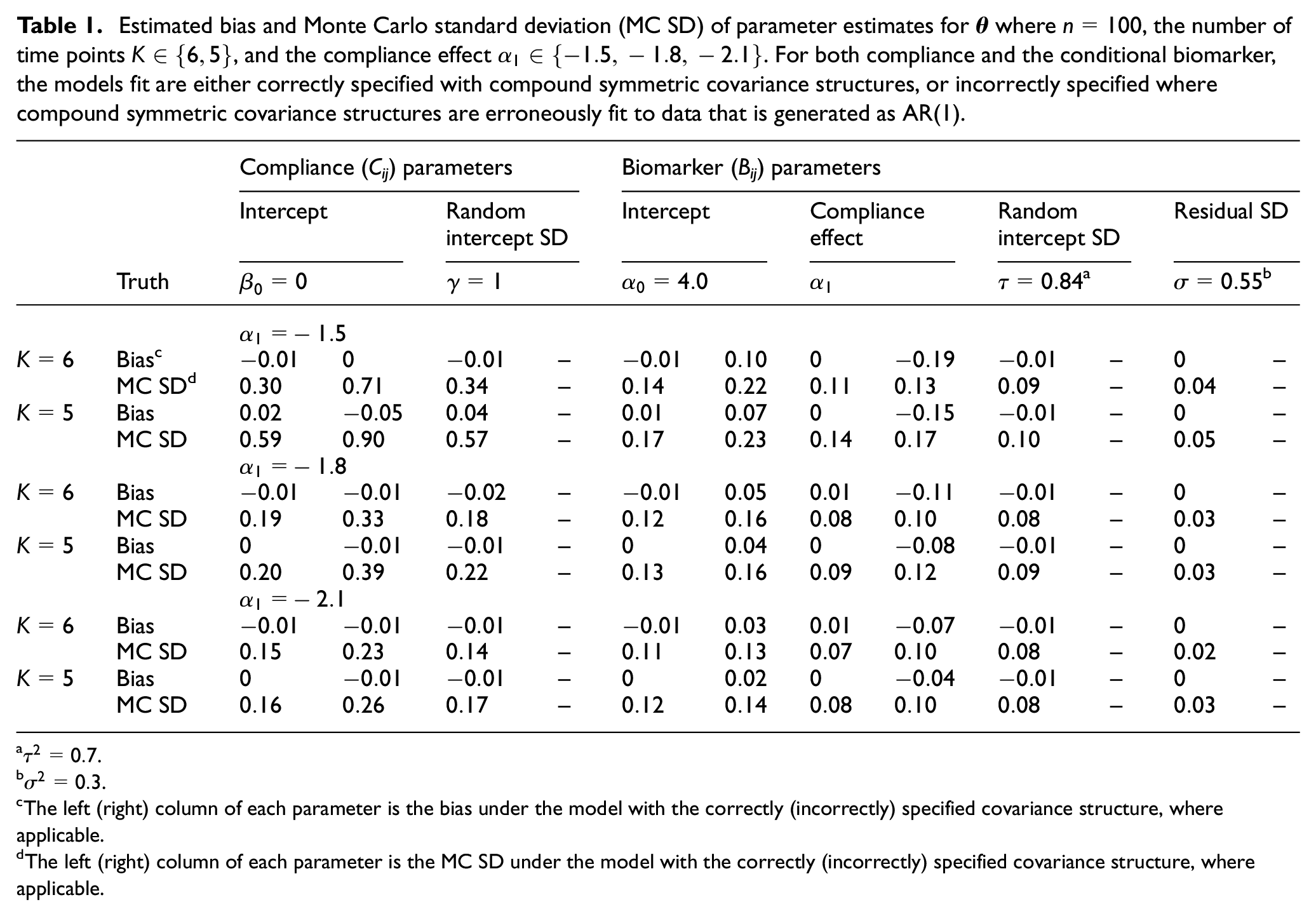
The left (right) column of each parameter is the bias under the model with the correctly (incorrectly) specified covariance structure, where applicable.
The left (right) column of each parameter is the MC SD under the model with the correctly (incorrectly) specified covariance structure, where applicable.
Across
True AUC values for
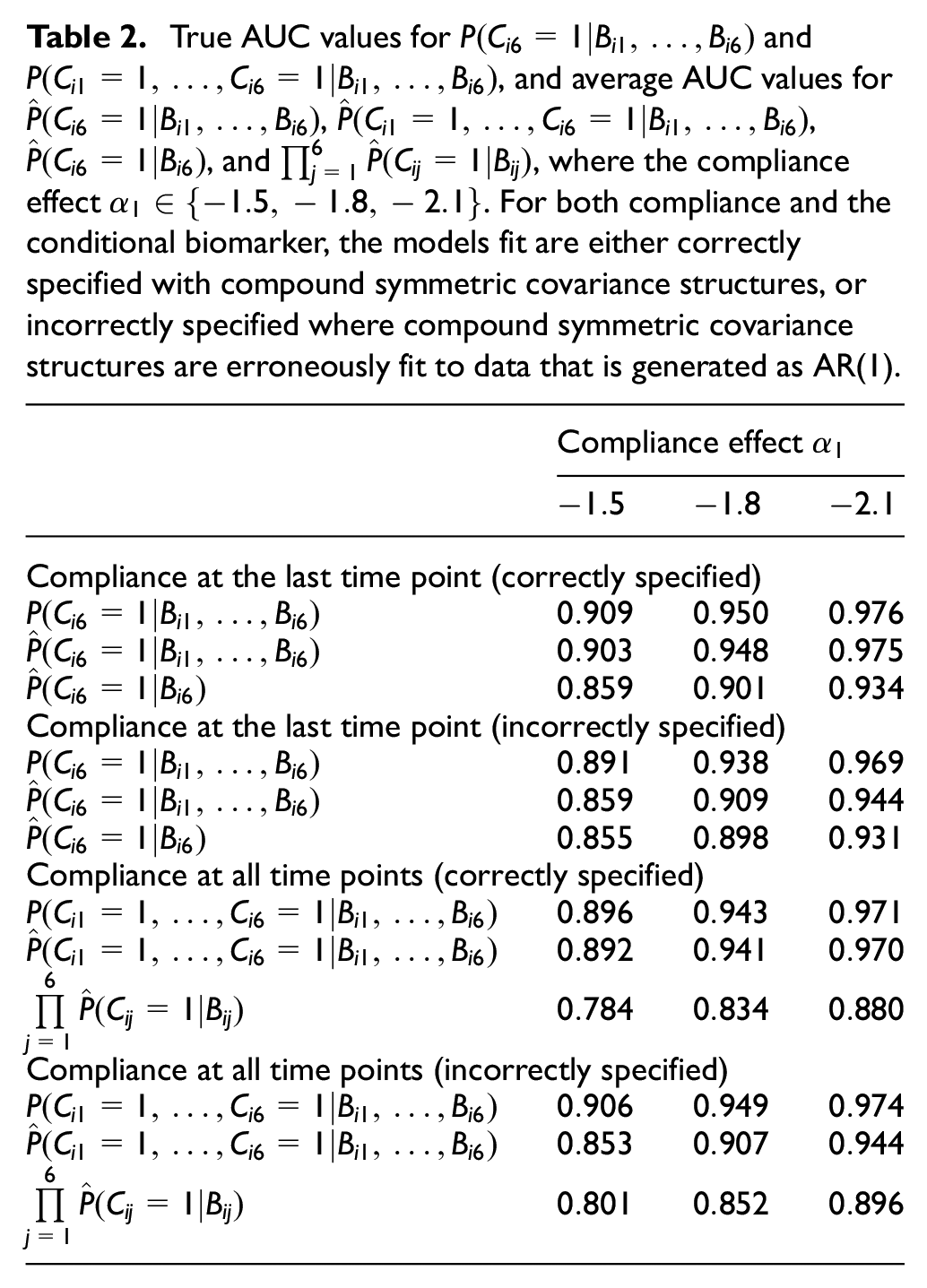
Comparing the compliance probabilities with those from Boatman et al.’s method, we found that
For prediction of compliance at the last time point, Figure 1 displays the (gray) calibration lines of
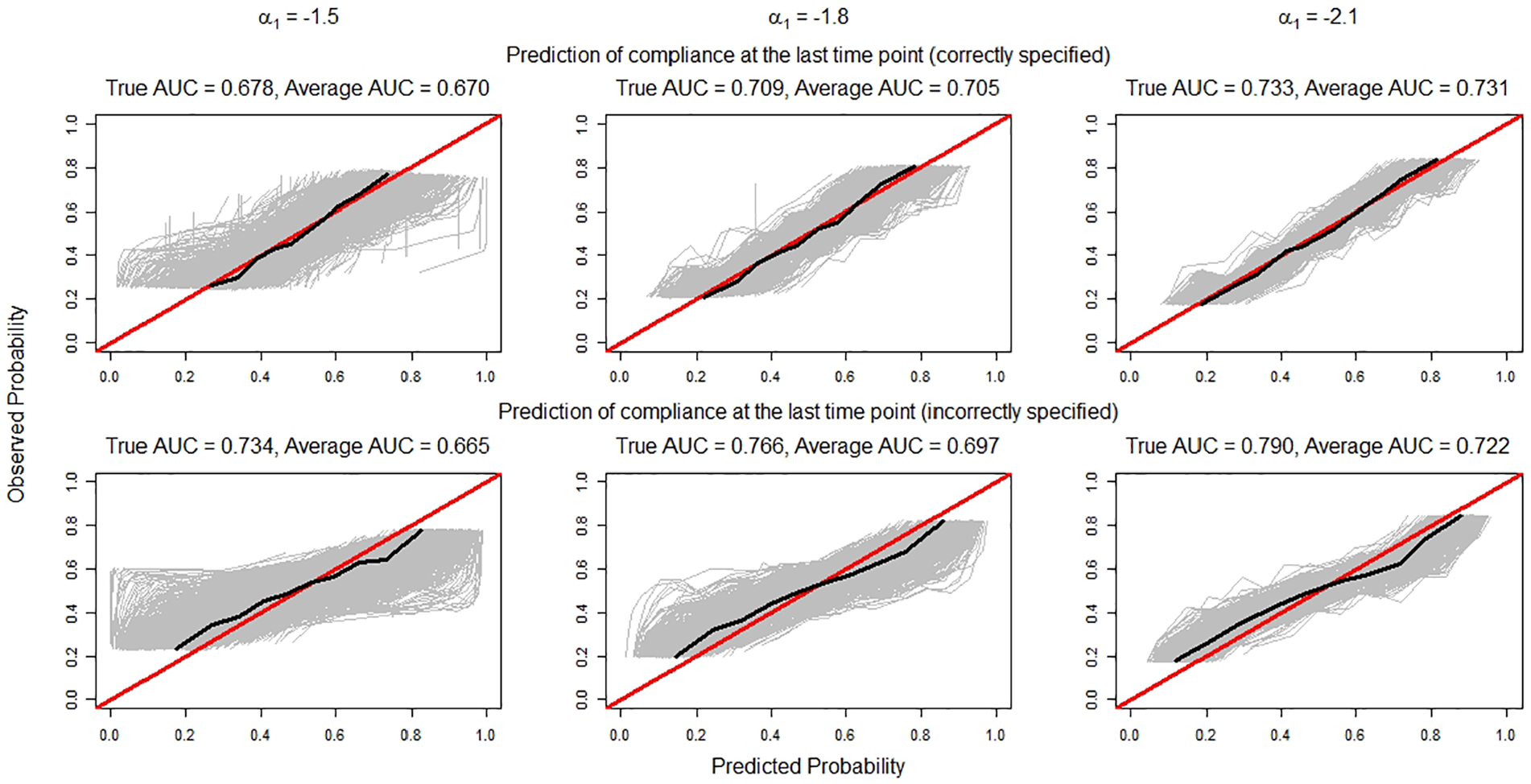
Calibration lines (in gray) for
Application to CENIC-P1S2
The model with a single compliance intercept for all time points returned a lower BIC (1882.35 vs 1902.72) compared to the model with a different compliance intercept for each time point; therefore, we assumed the more parsimonious model. Table 3 displays the parameter estimates and corresponding standard errors for
Parameter estimates and standard errors (SE) for
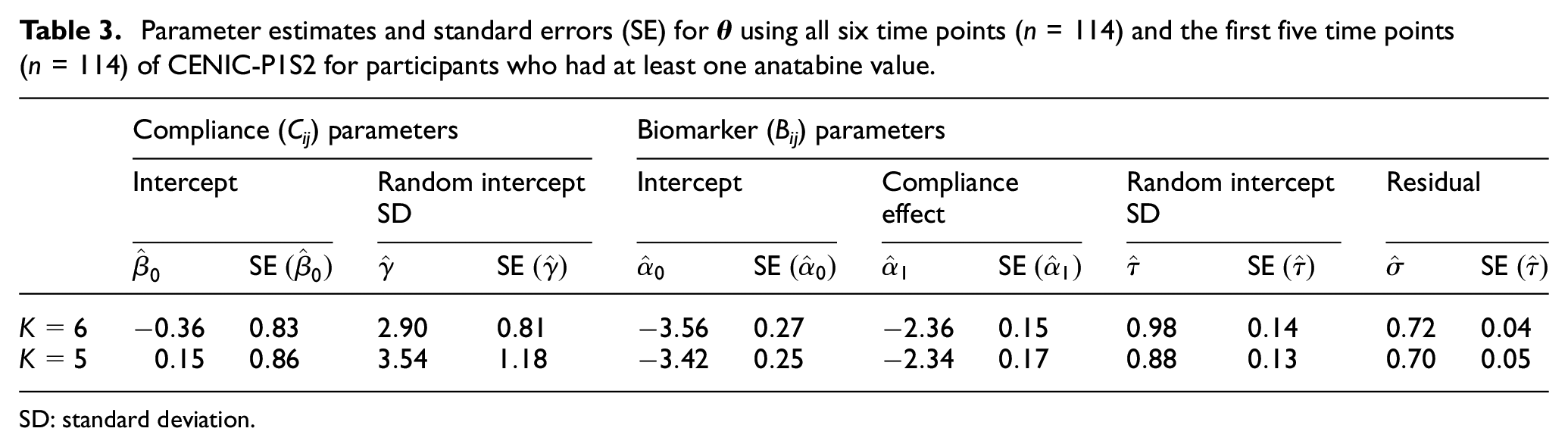
SD: standard deviation.
Figure 2 displays the spaghetti plot of log(anatabine) for the time points when participants had data for CENIC-P1S2. For the anatabine values, we find that consistency is as important as magnitude in determining the probabilities of compliance at all time points. Our method assigned high probabilities to participants who always had relatively low anatabine values. Conversely, our method assigned low probabilities to participants who had low anatabine values at some time points but upticks at others.
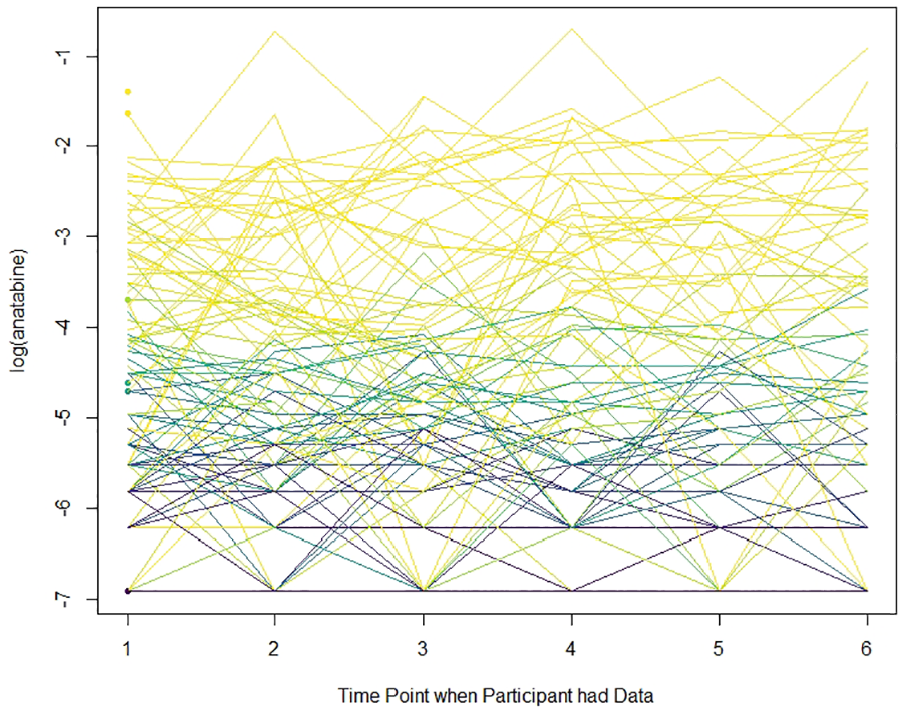
Spaghetti plot of the longitudinal biomarker for the time points when participants had data for CENIC-P1S2. Lines are shaded by the probability of compliance at all time points. Lighter lines indicate a probability closer to 0 while darker lines indicate a probability closer to 1. The x-axis refers to the time points when participants had data. For example, a participant with data only at time points 1 and 4 would have a line from 1 to 2.
For compliance at the last time point, the AUC of
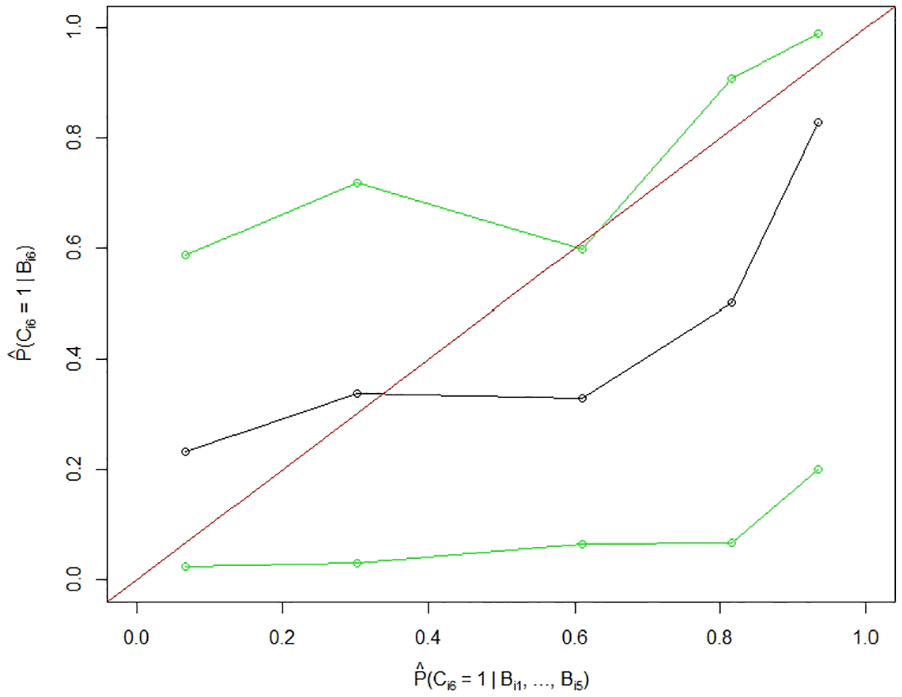
Averaged estimated prediction probability of
Discussion
For trials in which there is no known gold standard for measuring compliance, we have developed a method that can use longitudinal biomarker data to more accurately identify compliant participants, as well as predict which participants may or may not comply in the future. Our simulation study confirms that our method, which conditions on participants’ full biomarker history, is better able to discriminate between compliers and noncompliers compared to Boatman et al.’s method which conditions only on the most recent biomarker value. We also find that our prediction of compliance at a future time point conditional on biomarker data from previous time points is well-calibrated. Furthermore, when the covariance structures of both models for compliance and the biomarker are misspecified, we find that our method still has high discrimination. These results held across a range of differences in the biomarker between compliant and noncompliant participants.
In the application to the biomarker data collected from the
As demonstrated with the inclusion of the non-longitudinal hotel dataset, our method can include information from a partial gold standard. When compliance is known at all time points for a subset of participants, both longitudinal and non-longitudinal biomarker data can be incorporated into the biomarker log likelihood with the corresponding observed compliance patterns. Moreover, it is straightforward to adapt our method when compliance is known at some time points for a subset of participants. As only a subset of compliance patterns would be plausible, a mixture density with fewer compliance patterns can be fit for these participants.
For completed trials, our method can be used to improve estimation of causal effects. For example, the probability of compliance at all time points can be used to modify an inverse probability of compliance weight to more accurately weight the study outcomes, similar to Boatman et al. 11 For trials in progress, the prediction probability can be used to identify noncompliant participants to enable remedial intervention. This could both improve study outcomes and reduce the number of dropouts, the latter of which may also reduce the sample size at the end of the trial. That is, with fewer dropouts, fewer additional participants may be needed to maintain statistical power.
Although our method belongs to the broader class of growth mixture models commonly found in the psychometrics literature, 31 there are some key differences that distinguish our method. Specifically, the number of latent groups we considered (26 = 64) is substantially greater than in more traditional applications where 2–4 latent groups are more common. When the number of latent groups is small, the (marginal) probability of group membership is generally treated as a free parameter and the longitudinal data trajectory within each group is generally unconstrained. Given the large number of latent groups in our study, however, we constrained the probabilities of the latent groups by assuming mixed effects models for both compliance and the biomarker. Under this relatively simple modeling structure, the number of parameters is kept small relative to the number of latent groups.
Although we considered relatively straightforward models for both compliance and the biomarker, our method can be extended to fit richer longitudinal models with more parameters. Our method can also be adapted to a measure of compliance consisting of more than two levels, but substantially more computational power would be required to fit the models. That is, with more levels of compliance the number of compliance patterns would exponentially increase.
Our method does have some limitations. First, although mixed effects models can handle data which are missing at random, this assumption may not be realistic when studying compliance. Second, we have defined compliance to be a discrete random variable. Adapting our method to a continuous measure of compliance is not straightforward. Third, the compliance effect on the biomarker must be substantial to be reliably detected and larger samples are required to detect smaller compliance effects. Fourth, although likelihood-based tests and information criteria can help identify the optimal model structure, we have not developed model diagnostics specific to our method. Fifth, fitting mixed effects models to both observed and unobserved variables is computationally intensive and parameter estimates may be sensitive to initial starting values. Finally, as compliance is unobserved, it remains difficult to evaluate the discriminative performance of our method for clinical trials in practice. Model-based approaches can be used as in the applied example to CENIC-P1S2, but the AUC will likely be marginally overestimated.
In future clinical trials where a biomarker is known to respond to deviations from the treatment assignment, our method can be used to identify noncompliant participants both during and after the trial. The mode of noncompliance is immaterial; provided that noncompliance has a systematic effect on the biomarker, our method can be used for both participants who do not take any treatments and those who take alternatives. With this novel statistical method, trial designers should be better able to both adjust for noncompliance and prevent it from happening.
Supplemental Material
Compliance_Paper_Clean_3rd_Submission_Supp – Supplemental material for Detecting participant noncompliance across multiple time points by modeling a longitudinal biomarker
Supplemental material, Compliance_Paper_Clean_3rd_Submission_Supp for Detecting participant noncompliance across multiple time points by modeling a longitudinal biomarker by Ross L Peterson, Joseph S Koopmeiners, Tracy T Smith, Sharon E Murphy, Eric C Donny and David M Vock in Clinical Trials
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Heart, Lung, and Blood Institute (award number T32HL129956), the National Cancer Institute (award numbers R01CA214825 and R01CA225190), the National Institute on Drug Abuse (award numbers R01DA046320, R03DA041870, and U54-DA031659) and National Center for Advancing Translational Science (award number UL1TR002494). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Food and Drug Administration.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.