
Abstract
Introduction:
Clostridiodes difficile infection is the leading cause of infectious diarrhea in the United States, with substantial morbidity and mortality. Recurrent infection is especially challenging, with each recurrence increasing the likelihood of a successive recurrence, leading to cycles of prolonged symptoms, frequent antimicrobial use, and decreased quality of life. Fecal microbiota transplantation to prevent recurrent infection is a promising intervention with a large effect size in observational studies, but with conflicting results from randomized controlled trials. We are conducting a Veterans Affairs-wide randomized controlled trial utilizing centralized case identification, with enrollment and fecal microbiota transplant administration occurring at the participant’s home. This type of trial design significantly improves trial efficiency, greatly decreases trial cost, increases consistency of trial administration, and most importantly makes nationwide clinical trials in less-common diseases possible.
Methods:
This is a randomized comparison of capsule-delivered fecal microbiota transplant for the prevention of recurrent Clostridiodes difficile infection, administered after successful initial treatment of recurrent C. difficile infection with standard therapy. The primary endpoint is the incidence of recurrent C. difficile infection or death. Cases are identified by searching the Veterans Affairs Corporate Data Warehouse, with central study coordinators then reaching out to potential participants. Individuals meeting inclusion criteria and interested in participation are scheduled for in-home consent, randomization, and capsule administration, followed by telephone follow-up for 6 months. To mitigate risks of COVID-19, enrollment via video visits has been implemented.
Results:
A total of 102 participants have been enrolled through January 2021. Centralized case identification and in-home enrollment has facilitated enrollment from 34 unique states, with 38% being from rural or highly rural areas.
Discussion:
Centralized case identification and in-home enrollment is a feasible and innovative method of conducting randomized controlled trials in the Veterans Affairs system, improving access to clinical research for populations who may have difficulty engaging with the traditional model of clinical trials where enrollment is based at large hospitals in major metropolitan areas.
Keywords
Introduction
Clostridiodes difficile infection has transformed over the past 20 years from a relatively rare cause of antibiotic-associated diarrhea to the leading cause of infectious diarrhea in hospitalized patients, rivaling methicillin-resistant Staphylococcus aureus as the leading cause of all nosocomial infections.1–3 Nationwide estimates of yearly C. difficile infection exceed 450,000, resulting in 29,000 deaths and over $4 billion in healthcare costs.1,2,4 This increase in C. difficile infection has been even more striking in the Veterans Affairs Healthcare System, with rates of C. difficile infection exceeding those from non-Veterans Affairs facilities by two- to threefold. 5 Treatment guidelines from the Infectious Diseases Society of America 6 suggest that either vancomycin or fidaxomicin be used for the treatment of the initial episode of C. difficile infection, with vancomycin recommended for severe, complicated, or fulminant disease. Unfortunately, although initial clinical cure is achieved in >90% of patients with each of these drugs, recurrent C. difficile infection (variously defined as an episode of C. difficile infection that occurs 8 weeks or less after a previous episode, 7 or within 90 days of a previous episode provided that symptoms from the earlier episode resolved8,9) occurs in 15%–30% of cases.9–11 Patients with a first recurrence have a 40% risk of a second recurrence, after which the risk of subsequent recurrences exceeds 50%, leading to malnutrition, repeat clinic visits and hospitalizations, toxic megacolon and colectomy, and decreased quality of life. 12
The optimal treatment of recurrent C. difficile infection is unknown. Guidelines suggest that vancomycin or fidaxomicin should be used for the first recurrence. 6 Fecal microbiota transplant has emerged as a promising treatment for recurrent C. difficile infection in patients who have not responded to standard therapy. In 2013, a small unblinded trial of fecal microbiota transplant versus two vancomycin-based control groups was published, 13 reporting a success rate of 81% among the 16 participants randomized to fecal microbiota transplant, versus 23% and 31% in the two control groups. A systematic review of 21 case series and two randomized clinical trials 14 identified a total of 520 patients with recurrent C. difficile infection treated with fecal microbiota transplant. Although the overall rate of symptom resolution without recurrence was 85%, the majority of included studies were case series, and only one of the two randomized controlled trials had a non-fecal microbiota transplant control group. 13 A subsequent randomized controlled trial showed superiority of donor fecal microbiota transplant compared with administration of the participant’s own stool, 15 whereas another trial of fecal microbiota transplant versus tapered vancomycin was terminated early due to futility. 16
With the high burden of C. difficile infection in the Veterans Affairs system, high recurrence rates, and uncertainty regarding both how to treat recurrent C. difficile infection and the efficacy of fecal microbiota transplantation, a definitive randomized controlled trial of fecal microbiota transplant for the prevention of recurrent C. difficile infection was designed. Our aim was to conduct a randomized, blinded clinical trial of capsule-administered fecal microbiota transplant versus placebo, both administered after successful initial treatment of recurrent C. difficile infection with standard antimicrobial therapy, for the prevention of subsequent recurrent C. difficile infection in Veterans across the United States. The study chose a novel design of central identification, administration of the study intervention at the participant’s home, followed by phone follow-up conducted by a centralized study site. A traditional multi-site clinical trial seeking to cover the same population would have required 20–30 sites with local study teams. This would have created a large administrative burden, incurred at least twice the total cost, and still would have prevented Veterans receiving care at smaller facilities without research infrastructure from participating. Herein we describe the novel study design, which was adapted to accommodate COVID-19 challenges, and an interim update of recruitment to date.
Methods
Study information is posted online at https://clinicaltrials.gov/ct2/show/NCT03005379 and is summarized below.
Study setting
This is a randomized, double-blind, placebo-controlled trial of capsule-delivered fecal microbiota transplant for the prevention of recurrent C. difficile infection, administered after successful initial treatment of recurrent C. difficile infection with standard antimicrobial therapy. The study was approved by the Human Rights Committee at the West Haven Veterans Affairs Cooperative Studies Program Coordinating Center and by the Institutional Review Board at the Veterans Affairs Minneapolis Healthcare System. It is funded and administered by the Veterans Affairs Cooperative Studies Program. The study has one central site located at the Veterans Affairs Medical Center in Minneapolis, Minnesota, where the two study co-chairs and the national study coordinator are employed, with four central study coordinators. We are open to recruitment of all eligible veterans across the United States.
Study population
Identification and contact of potentially eligible veterans
Eligible participants are Veterans who have had one or more episodes of recurrent C. difficile infection (defined as >3 loose/watery stools/24 h for 2 consecutive days with a laboratory confirmation of C. difficile and have received C. difficile infection treatment).
The study design effectively leverages the Veterans Health Administration’s national electronic medical record storage system to identify and recruit Veterans across the United States. We have developed and validated algorithms to identify all C. difficile infection testing results from the Veterans Affairs’ Corporate Data Warehouse, a centralized repository of all Veteran’s Health Administration medical, laboratory and pharmacy records that receives data daily from every Veteran’s Health Administration facility, encompassing over 8 million Veterans. An incident case of C. difficile infection is identified through electronic monitoring of orders for C. difficile testing, lab results, diagnostic codes for outpatient and inpatient visits, and prescriptions. Our algorithm also identifies individuals with two positive laboratory C. difficile infection tests within 90 days of each other (key study inclusion criteria). The study database is subsequently populated with these names, and central study coordinators screen cases found through this automated system using the patient’s electronic medical record to assess for inclusion/exclusion criteria using national Computerized Patient Record System access. Information on rurality is based on the Veterans Affairs administration’s definition that uses the rural–urban commuting areas system. Areas are defined as Urban if at least 30 percent of the population resides in an urbanized area, Rural Areas are land areas not defined as urban or highly rural, and highly Rural Areas are sparsely populated areas in which less than 10% of the working population commutes to any community larger than an urbanized cluster, and are typically towns of no more than 2500 people.
Veterans that appear eligible are contacted through their treating healthcare provider and subsequently sent a letter of invitation, which includes information about the study and an option to opt out if they do not wish to be contacted further. Seven days after the mailing of the letter, Veterans who did not opt out are contacted via telephone. The study coordinators also contact the Veteran’s provider and ask the provider or a member of their care team to obtain permission from the Veteran to initiate telephone contact, thereby allowing contact before the 7-day opt-out period. The study coordinators also receive direct referrals from providers who have become aware of the trial. Veterans with recurrent C. difficile infection who (1) have completed a standard course of antimicrobial therapy with a clinical response of resolution of symptoms, (2) express interest in enrolling, and (3) meet inclusion criteria are scheduled for a visit with a central study coordinator within the enrollment window (i.e. 2–14 days after the conclusion of their standard antimicrobial therapy). Utilizing methods that Veterans Affairs homecare providers use prior to all new homecare visits, study personnel review the medical records to ensure there are no safety risks for the visiting coordinator.
Recruitment at home
Before committing to travel, the central study coordinators contact the potential participant to verify that they have completed their standard antimicrobial therapy, that they are still planning to enroll, and that they have met the criteria of having resolved/improved symptoms of C. difficile infection (3 or fewer stools per day for at least 2 days, without subsequent worsening, and no other manifestations of C. difficile infection such as ileus or toxic megacolon). If all these conditions are met, the study coordinators travel to the Veteran’s residence on a mutually agreed-upon date and time, with the randomly allocated treatment. Once at the participant’s home, the coordinator reviews the study information with the participant, obtains written informed consent, and proceeds with treatment administration (oral administration of the randomly assigned treatment capsules), data collection, and collection of an optional rectal swab for microbiome analysis. Following capsule administration, follow-up is done via scheduled telephone calls from study personnel, with ad hoc contact triggered either by participants reporting symptoms or study personnel noting clinical manifestations of recurrence in the medical record, which is monitored on an ongoing basis for the 6-month study period.
To enhance recruitment, we have disseminated information about the trial to Veterans Affairs Primary Care, Infectious Disease, and Gastrointestinal physicians. To address concerns regarding participant and research coordinator safety brought on by the COVID-19 pandemic, we added virtual enrollment via Veterans Video Connect as an additional platform for enrollment. This allows remote recruitment, thus obviating potential risks associated with study personnel travel and at the same time, avoiding any potential risk of SARS CoV-2 exposure to Veterans. Shifting enrollment activities to such virtual visits was explicitly endorsed by the Veterans Affairs Office of Research and Development to mitigate the risks of conducting clinical research during the COVID-19 pandemic. After a hiatus of approximately 5 months to gain approvals of the addition of a remote enrollment strategy, the study resumed enrollment. During this time, study follow-up of existing participants and other study activities were not interrupted.
Study design
Inclusion and exclusion criteria
Study inclusion criteria are adults enrolled in a Veterans Healthcare Facility with one or more episodes of recurrent C. difficile infection defined as either >3 loose/watery stools/24 h for 2 consecutive days with C. difficile infection treatment, and not explained by another diagnosis AND laboratory confirmation of C. difficile; or ileus, or toxic megacolon AND laboratory confirmation of C. difficile, either occurring within 90 days of a prior C. difficile infection episode with similar symptoms and laboratory confirmation. Individuals must have resolution or improvement of symptoms from the most recent C. difficile infection episode, for a 48-h period during treatment and be able to be randomized within the enrollment window, which is between 2 days after completion of antimicrobial therapy for C. difficile infection (to allow for a washout period) to 14 days after completion of therapy or 30 days after the onset of C. difficile infection, whichever is later. The exclusion criteria are inability to swallow capsules, pregnancy or planning to be pregnant, breastfeeding, receipt of cytotoxic chemotherapy, intravenous or subcutaneous immune globulin, or confirmed neutropenia (absolute neutrophil count of <1000 cells/µL) within the past 3 months, inflammatory bowel disease or other chronic diarrheal disease/fecal incontinence predating C. difficile infection, ongoing antibiotic use other than those for the current episode of C. difficile infection, prior fecal microbiota transplant, life expectancy of <8 weeks, anaphylactic food allergy, active enrollment in another research study on antibiotics, probiotics, or fecal microbiota transplant without investigators approval, presence of an ileostomy or colostomy, human immunodeficiency virus with CD4 count <200 cells/µL in prior 3 months, bone marrow/peripheral blood stem cell transplant in the past year, decompensated cirrhosis and unlikely to follow study protocol.
Study intervention
The experimental intervention of the study is capsule-delivered fecal microbiota transplant. Oral capsule and colonoscopy administered fecal microbiota transplant appear to have similar efficacy. 17 The source of the fecal microbiota transplant capsule is the University of Minnesota, which maintains a robust fecal microbiota transplant program. The dose of fecal microbiota transplant is based on extensive clinical and research experience using a highly screened and selected pool of donors.15,18–21 Treatment is administered in 5 double-encapsulated size 00 enteric capsules, taken in one sitting lasting no more than 90 min, containing a total of at least 5 × 1011 microbes freeze-dried using trehalose as a cryoprotectant. The manufacturer is also supplying identical placebo capsules, both of which are shipped at −80 C to a centralized research pharmacy that catalogs the bottles and sends them to the Minneapolis Veterans Affairs Medical Center for short-term storage and dispensing.
Study endpoints
The primary endpoint is the incidence of recurrent C. difficile infection (definite or possible) or death within 56 days of randomization among participants receiving fecal microbiota transplant versus those receiving placebo capsules. Definite C. difficile infection recurrence is defined as new onset of more than 3 loose or watery stools in 24 h for 2 consecutive days with laboratory confirmation of a positive C. difficile toxin test from a stool specimen in the absence of other diagnosis; or ileus, toxic megacolon, or colectomy with a positive toxin test. Possible recurrence is defined identically as above but without laboratory confirmation of C. difficile toxin being present (no specimen, not tested, test not directly detecting toxin, or negative toxin test). Secondary endpoints include the incidence of recurrent C. difficile infection (definite or possible) or death within 6 months of randomization, the incidence of definite recurrent C. difficile infection within 56 days after randomization, the incidence of possible recurrent C. difficile infection within 56 days after randomization, the incidence of diarrhea that is negative for C. difficile (both by toxin and polymerase chain reaction) laboratory testing within 56 days after randomization, self-reported quality of life within 56 days after randomization, the number of C. difficile infection recurrences within 6 months after randomization, and to compare the incidence, severity, and relatedness of serious adverse events and complications within 6 months of randomization. In addition, there are several exploratory aims to characterize the stool microbiome and compare diversity and flora at various time points during the study between the two groups. A stool sample for the assessment of the colonic microbiome will be requested at the time of randomization, and again 14 and 56 days after randomization.
Analysis plan
The primary outcome (recurrent C. difficile infection or death within 56 days) will be compared between the two arms using a two-sided Type I error of 0.05. The incidence of secondary outcome of recurrent C. difficile infection or death within 6 months will be analyzed with generalized linear model. Kaplan-Meier survival curves, adjusted for censoring due to loss to follow-up, will also be used to evaluate treatment effects for 6 month recurrent C. difficile infection.
Target sample size and randomization
We anticipate a rate of recurrent C. difficile infection of 35% following standard antimicrobial therapy. Fecal microbiota transplant is expected to reduce the incidence of recurrent C. difficile infection to 20% (an absolute difference of 15%). With a target sample size of 390 participants and the above assumptions, the study will have 90% power to detect a 15% absolute difference (20% vs 35%) with a two-sided significance threshold of 0.05, and a 5% dropout rate.
All eligible participants are randomized in a 1:1 ratio to the two treatment arms. Randomization is permuted block, stratified by number of C. difficile infectionepisodes (1or >2) and is performed using a randomization system accessed at the study website using a computer-generated randomization scheme, thereby concealing the allocation. All study personnel and participants are blinded to the treatment assignment. The randomization process starts with the study coordinator filling out a randomization form, getting a bottle number from the system, confirming eligibility criteria with the prospective subject via phone. In the next subsequent days, the study coordinator will travel to the potential participant’s home, obtain written consent, including detailed instruction in the follow-up protocol, and then administer the randomly assigned treatment. Following the visit, the study coordinator confirms through the randomization system the successful completion of that randomization. If participants change their mind and/or administration of the study therapy does not occur, the study coordinator will report to the system that the potential participant was not successfully randomized and closes out that randomization instance. Only successfully randomized patients are counted and followed as study participants. Up until the end of January 2021, a total of 11 patients (8 during travel phase and 3 during video visit) were not successfully randomized.
Study randomization visit
Participants are mailed the consent form prior to the visit and instructed to read it thoroughly. Once they sign and return the consent form, the study capsules are mailed to them. All details of the visit are discussed with the potential participant on the phone, including confirming that participant remains eligible, the day prior to the visit. At the visit, participants are observed taking the medications (5 capsules) and monitored for 2 h after.
Study follow-up
Study follow-up is done by telephone. Participants are assessed for symptoms of C. difficile infection and any treatment-related adverse events with a call from study personnel at days 2, 14, and 56 after capsule administration. The day 56 (8 week) call is used to assess the primary endpoint, confirmed or possible recurrent C. difficile infection, and the secondary endpoints, including a quality-of-life questionnaire. We collected information on any adverse events in the first 14 days, and potentially related adverse events and serious adverse events for the full 6 months.
Participants experiencing new diarrhea, in addition to seeking clinical care at their local facility, were instructed to contact the central study coordinator, collect a stool sample using a collection kit provided by the central study coordinator, and mail the sample in a pre-paid temperature-controlled mailer to the central laboratory where it is tested for C. difficile toxin by Enzyme Immunoassay and the presence of the toxin B gene by polymerase chain reaction. The temperature-controlled mailers were tested using temperature recording devices. Results from four test shipments indicated that the temperature remained under 8.5°C for 6 days. At 6 months, a final call by the central study coordinator occurs to assess for any new symptoms of recurrent C. difficile infection, serious or potentially related adverse events, and any other reported symptoms.
Results
The study began recruiting in October 2018 and has recruited 102 participants to date (January 2021). The eligibility screening algorithm has identified an average of 15 participants per week. The central study coordinators have reviewed 2050 charts for eligibility (Figure 1). Of the 102 individuals enrolled, 87% are male, 88% White and 6% Hispanic (Table 1). The enrolled individuals are from diverse geographic locations including 34 U.S. states, with the largest numbers being from Texas, Florida, California and New York (Figure 2). Nearly 38% live in a rural or highly rural area, and the average distance to their nearest Veterans Affairs facility is 46.6 miles. Novel aspects of this study have also resulted in some challenges, including two instances of Veterans changing their minds about participation after the central study coordinator had already traveled, and another instance of an exclusionary criterion only noted when the coordinator was performing a final check before randomization, resulting in the Veteran not being randomized. Of participants experiencing symptoms of recurrence, 19 out of 32 have sent in a sample to the central laboratory and all samples were adequate. Seventy-six participants have reached the 56-day endpoint.
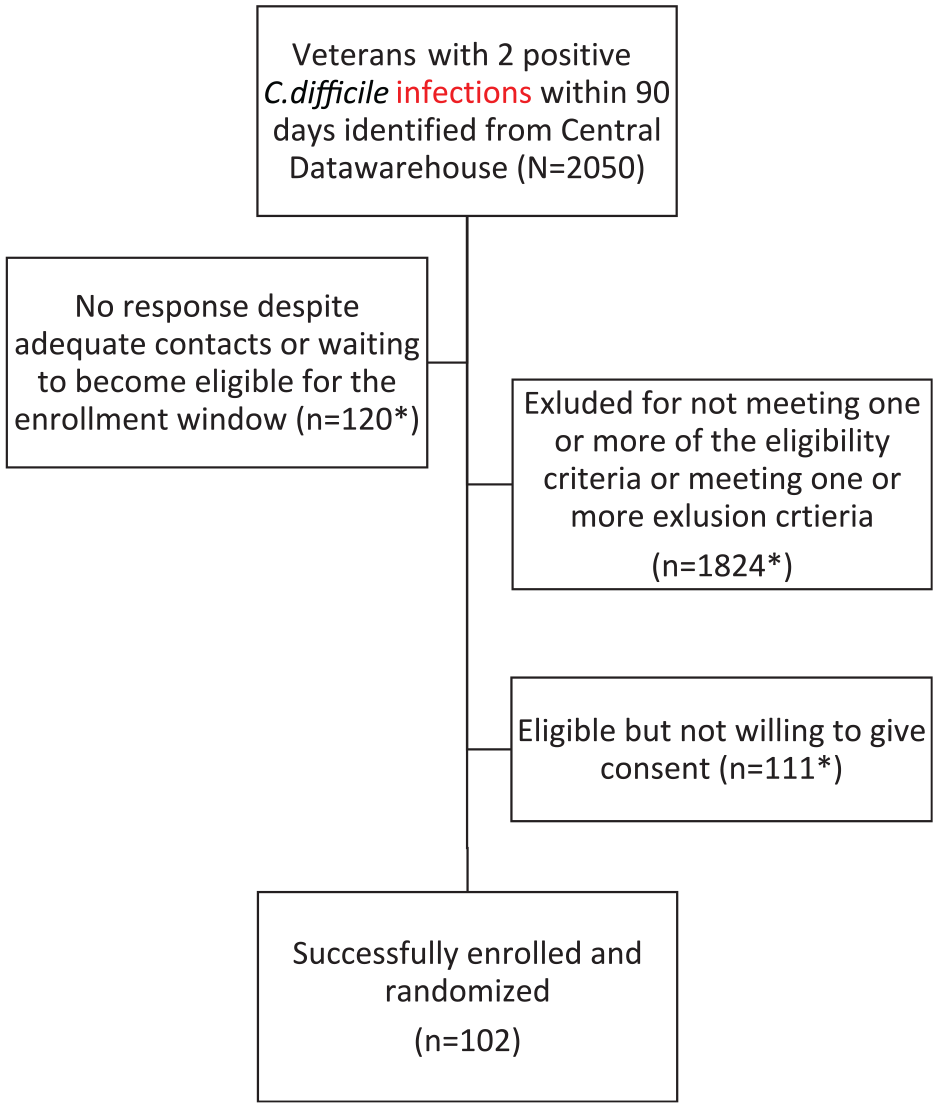
Flowchart of enrollment.
Demographic characteristics of enrolled individuals (n = 102).
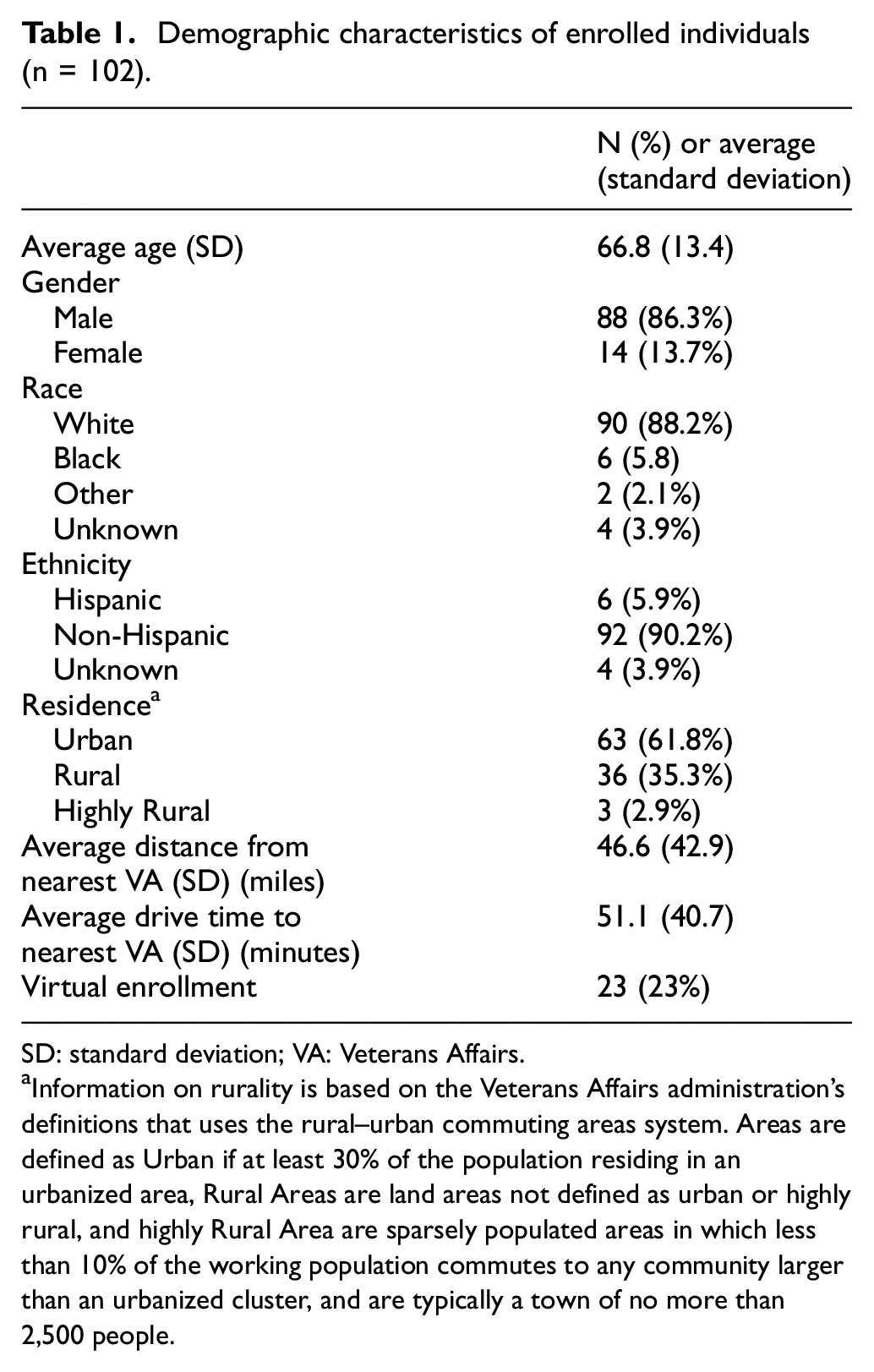
SD: standard deviation; VA: Veterans Affairs.
Information on rurality is based on the Veterans Affairs administration’s definitions that uses the rural–urban commuting areas system. Areas are defined as Urban if at least 30% of the population residing in an urbanized area, Rural Areas are land areas not defined as urban or highly rural, and highly Rural Area are sparsely populated areas in which less than 10% of the working population commutes to any community larger than an urbanized cluster, and are typically a town of no more than 2,500 people.
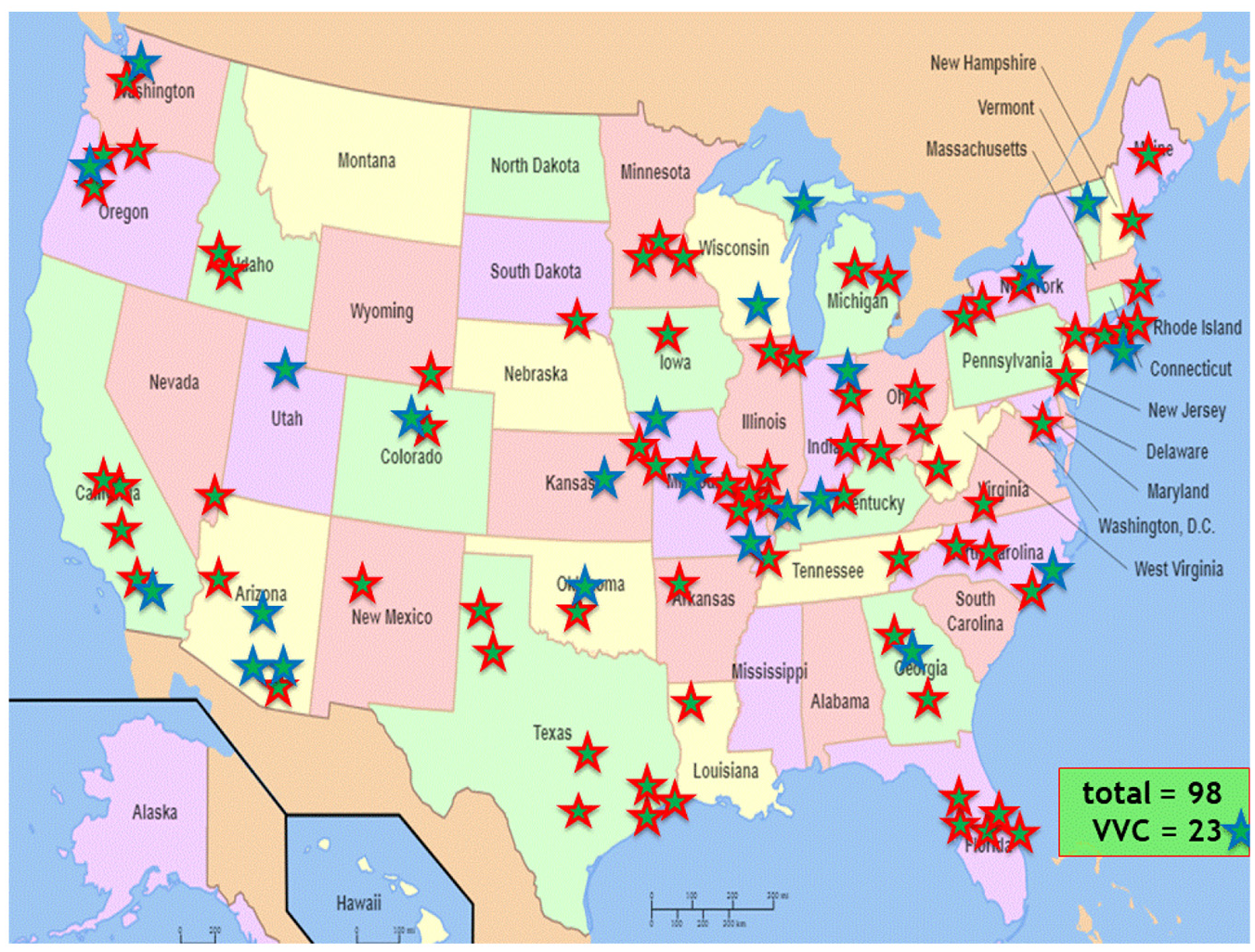
Geographic location of the enrolled individuals across the United States.
Remote enrollment with Veterans Affairs Video Connect
The study is currently enrolling with the added option of remote enrollment and consent via video visit, followed by shipment of study pills to the participant and video observation of administration of the study capsules. Twenty-three participants have enrolled virtually thus far.
Discussion
Recurrent C. difficile infection is a challenging dilemma for clinicians. Although effective treatments are available for C. difficile infection, 15%–30% of patients who respond to initial courses of treatment develop recurrent C. difficile infection, usually within 1–4 weeks of completing treatment with either vancomycin or metronidazole. 12 The risk of recurrent C. difficile infection is even greater in patients who have already had one or more recurrences, and many patients develop repeated episodes that may continue to occur over a period of months or years. Treatment of recurrent C. difficile infection is a priority for the Veterans Affairs Healthcare Administration. Using a unique study design and novel recruitment methods, our study aims to answer important questions about efficacy and safety of fecal microbiota transplant which will inform guidelines and determine policy on fecal microbiota transplant within Veterans Affairs healthcare and beyond.
Two important features of this study are the centralized nature of potential participant identification and the delivery of the study intervention at the Veteran’s home. The centralized identification utilizes the Veterans Affairs Administration’s robust data infrastructure and local programming skills to allow Veterans Affairs–wide patient identification quickly and precisely which allows study coordinators to focus on actual patient recruitment. The ability to travel to the Veteran’s home brings the possibility of participating in clinical research to Veterans across the United States, instead of just those enrolled in or close to the sort of large tertiary medical centers which are the usual sites for multi-center trials. Central identification with travel to the Veteran’s home also allows a nationwide study to be run with streamlined staffing and a single institutional review board oversight of study activities. Another advantage of this trial design is the cost savings. There are about 1300 recurrent C. difficile infections per year within Veterans Health Administration. With this number of cases, it is not cost effective to run a clinical trial with a usual setup at 25–30 medical centers. Our centralized case identification and travel model greatly reduced the budget and made the trial possible.
When the ongoing COVID-19 pandemic paused elective nationwide travel, we switched to a new model that utilizes the VA Video Connect platform to perform study recruitment when travel is not feasible or safe. VA Video Connect is currently in use for clinical visits between Veterans and staff. With the emergence of the novel coronavirus that causes COVID-19, the possibility of transmission of COVID-19 through travel or face-to-face meeting and the guidelines for social distancing have largely halted recruitment for clinical trials. Under these conditions, our study can proceed with consent via video visit and direct observation of participants taking the study capsules on video. The ability to enroll remotely obviates the need for travel entirely and allows for continuing recruitment. However, while VA Video Connect is available to all Veterans across the country, Veterans without access to the Internet via an electronic device such as a computer with camera, smart phone, or tablet may be excluded from enrollment. This has not come up so far in our enrollment experience, but theoretically poses a risk of lack of generalizability. Furthermore, the trial is limited to Veterans and generalizability to the general population is unknown.
Our preliminary results are encouraging in that eligible patients are being identified across the United States, with 33% of enrolled participants residing in areas designated as rural. Central study coordinators report no safety concerns utilizing the safety pre-screening methods used by home health providers.
In summary, we describe a novel study design using centralized case finding and delivery of the study intervention to the Veteran’s home whereby a nationwide study of fecal microbiota transplant versus placebo for the prevention of subsequent recurrent C. difficile infection is being carried out by the Veterans Affairs Cooperative Studies Program.
Footnotes
Acknowledgements
We thank our study staff; Ruth Anway, BA, RN, John Belew, BSN, Jacob Beste, BSN, Alex Pretti, BA, for their help with the study. This study was supported by the Cooperative Studies Program of the Department of Veterans Affairs Office of Research and Development. The contents do not represent the views of the U.S. Department of Veterans Affairs or the U.S. Government.
Planning committee members:
Minneapolis Veterans Affairs medical center Minneapolis MN
Aasma Shaukat MD MPH*
Dimitri M. Drekonja, MD MS*
Frank Lederle, MD (deceased)
Jason A. Dominitz, MD MHS, VA Puget Sound Health Care System, Seattle WA
Dale N. Gerding MD, Edward Hines Jr. Veterans Affairs Hospital, Hines, IL
*Denotes study Co-Chairs
West Haven Cooperative Studies Coordinating Center, West Haven, CT
John Concato, MD, Director of the VA Clinical Epidemiology Research Center, West Haven, CT
Jane Hongyuan Zhang, PhD
Tassos C. Kyriakides, PhD
Peter Guarino, PhD
VA Cooperative Studies Program Research Pharmacy, Albuquerque, NM
Anne Davis-Karim, PharmD
Stuart Warren, JD, PharmD
VA Central Office Research and Development
Grant D. Huang, MPH, PhD
Timothy O’Leary, MD, PhD
Dave Burnaska, MPA
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Veterans Affairs Cooperative Studies Program 2004 (NCT03005379).