
Abstract
Despite enormous advances in biomedical science, corresponding improvements in health outcomes lag significantly. This is particularly true in the United States, where life expectancy trails far behind that of other high-income countries. In addition, substantial disparities in life expectancy and other health outcomes exist as a function of race, ethnicity, wealth, education, and geographic location. A major reformation of our national system for generating medical evidence—the clinical research enterprise—is needed to facilitate the translation of biomedical research into useful products and interventions. Currently, premarket systems for generating and evaluating evidence work reasonably well, but the postmarket phase is disaggregated and often fails to answer essential questions that must be addressed to provide optimal clinical care and public health interventions for all Americans. Solving these problems will require a focus on three key domains: (1) improving the integration of and access to high-quality data from traditional clinical trials, electronic health records, and personal devices and wearable sensors; (2) restructuring clinical research operations to support and incentivize the involvement of patients and frontline clinicians; and (3) articulating ethical constructs that enable responsible data sharing to support improved implementation. Finally, we must also address the systemic tendency to optimize individual components of the clinical research enterprise without considering the effects on the system as a whole. Overcoming suboptimization by creating incentives for integration and sharing will be essential to achieve more timely and equitable improvement in health outcomes.
Keywords
Introduction
Biomedical science and technology are undergoing a remarkable period of discovery and development, one driven in large part by the innovations of U.S. scientists and engineers. Unfortunately, these advances are not resulting in superior health outcomes for U.S. patients and consumers. 1 However, if supported by effective policies and investment, the intersection of discovery in biomedical science, technology, and communication has the potential to usher in a new era of better health for the United States and the world.
Previous policies and infrastructure investments 2 provide a solid base upon which to build an effective system for evaluation and implementation across the spectrum of new biomedical product and technology development, pivotal clinical trials, and postmarket assessments. This improved system for generating and evaluating scientific evidence could better define when and how products, technologies, and behavioral interventions should be deployed, and when these interventions should be eschewed.
The U.S. Food and Drug Administration (FDA) has a critical role to play in reshaping the systems for evidence generation and dissemination. Its unique position as a regulatory, scientific, and public health agency gives it both insight and clout. The majority of clinical research funding comes from the medical products industry, 3 and FDA regulations and guidance provide a framework for the creation of scientific evidence that helps us better understand the balance of benefits and risks of medical products when used in practice. However, holistic solutions to broader issues currently impeding evidence generation will require collaboration across many sectors, together with creative and collaborative approaches to overcome financial disincentives that hinder common-sense improvements.
The “burning platform”
Given the prowess of U.S. biomedical science, continuing declines in life expectancy are particularly frustrating. Recent studies1,4 show U.S. life expectancy decreasing while gains continue in other high-income countries, with the difference between the United States and the average of other high-income countries now estimated to be a full 5 years. Some of this difference is driven by the response to the SARS-CoV-2 pandemic, as evidenced by higher rates of excess mortality and lower rates of vaccination in the United States than in other high-income countries. 5 However, most of it results from a combination of high rates of common chronic diseases such as heart disease and diabetes, substance use disorders, drug overdose, gun violence, and mental health issues leading to suicide.
These differences are not uniformly distributed. Disparities according to sex, gender, race, and ethnicity persist. 6 Other social determinants of health including socioeconomic status, employment, housing, education, and geography (which includes disadvantaged urban areas and large parts of rural America) are critical factors. Differences in life expectancy across U.S. counties now vary by more than 20 years—increasing in areas with high income and access to a concentration of resources (including university towns in rural states), while rural areas see rapidly deteriorating health status. 7
Addressing these stark gaps in outcomes is clearly an urgent priority for the United States. A key part of doing so requires continuous efforts to design more effective and efficient methods of evidence generation for evaluating medical products. The vast majority of new drugs and biologics fail in development because of off-target effects, failure to effectively engage the therapeutic target, or insufficient health effects despite target engagement.8–10 Many devices also fail in development because of design or engineering issues, or due to inadequate effects on the disease target. Current methods of evidence generation for premarket product development arguably are winnowing ineffective and unsafe medical products, thereby allowing safe and effective products to reach the market. However, the cost is enormous and continues to escalate. The high failure rate of the products themselves suggests that we need a more efficient system to enable more “shots on goal” to develop effective new products.
The major gap in our system of evidence generation occurs in the postmarket phase (and, potentially, in the late phase of premarket research for products that require large studies). During this phase, the system often fails to deliver evidence needed to confirm or refute the basis for accelerated product approvals, understand the benefits and risks of interventions in real-world practice, the comparative benefits and risks of interventions, and the data needed to calculate resource use that would allow the value of interventions to be estimated. Most clinical practice guidelines are not supported by high-quality evidence, 11 and the U.S. healthcare delivery system spends over $4 trillion per year for outcomes inferior to those of other high-income countries.12,13 These facts underscore the need for better evidence to support more use of effective interventions and to combat the use of interventions that are not effective in particular populations and indications.
In the face of poor health outcomes, health disparities, and evidence gaps, analyses reveal tremendous inefficiencies and inequities in a clinical research system that has been characterized as slow, expensive, and unreliable. Furthermore, it is not user-friendly either for research volunteers or clinicians who wish to participate in the research system. All these issues are magnified in the context of structural issues in our healthcare delivery and clinical research systems that have systematically disadvantaged minority populations, those with less education and wealth, and those living in rural areas.
Empirical evidence points to a continued escalation of costs in clinical research, 14 driven in large part by the costs of human labor. This is particularly true when the number of study procedures proliferate 15 and regulatory demands escalate with consequent demands on personnel.
Given the tremendous shortfall in evidence needed to guide optimal health interventions, coupled with systemic inefficiencies and rising costs, the need for structural change and realignment is obvious. The large gaps in the postmarket space, where the FDA generally has less direct influence on evidence generation, will require collaboration across multiple sectors. However, the agency’s remit does include an important component of the postmarket phase. The FDA mission statement includes “ensuring the safety, efficacy, and security of human and veterinary drugs, biological products, and medical devices” and “helping the public get the accurate, science-based information they need to use medical products and foods to maintain and improve their health.” 16 Thus, the FDA has a stake in a better evidence generation system that goes well beyond the premarket phase. However, developing such a system will require coordinated action across the clinical research enterprise.
The role of evidence
One could argue that the greatest impact on health outcomes would come from prioritizing the application of interventions known to be effective (as opposed to developing new technologies or investing in more evidence generation). However, too often we lack fundamental evidence to have confidence that we understand the full spectrum of benefits and risks of possible interventions. Although FDA approvals for marketing are based on convincing evidence about risk and benefit in a particular patient group for a specific intended use, use of medical products in practice almost always involves additional populations and uses. For example, by definition, the use of the accelerated approval pathway is based on unvalidated surrogate or intermediate clinical endpoints that require follow-on studies to verify the predicted clinical benefit.
Even when the evidence is clear, as seen with the vaccines developed and granted emergency use authorization for the prevention of COVID-19, a new product or technological intervention can have a limited impact on outcomes because of implementation failures. The potential value of high-quality evidence will only be realized when our implementation system is linked more effectively to following through on using evidence to guide effective practice. As an example, despite high-quality randomized trials and numerous real-world, non-interventional studies that demonstrate reductions in death and hospitalization with vaccination, many Americans have gone unvaccinated, and the majority have not received updated boosters, which are recommended based on the evidence.
A key tenet of the clinical research enterprise and regulatory authorities administered by agencies (including the FDA) that oversee it is that it is far preferable to test potential interventions proactively than to try to derive the evidence after deployment; this tenet is an essential element of FDA regulation. In other words, care should be taken to develop concepts for health interventions, generate high-quality, interpretable, and applicable evidence for or against them, and then focus on implementing interventions proven to be effective. This contrasts with a retrospective approach, whereby patients are exposed to interventions with unknown risks and uncertain benefits and researchers attempt to sort out the balance of risks and benefits after the fact through market forces and retrospective analysis.
Although this point might seem obvious, product development did not always follow the proactive path. Rather, the proactive evidence generation paradigm that underpins approval for regulated medical products evolved from a series of public health disasters involving the use of dangerous medical products by naive clinicians and patients. 17 Prior to the Kefauver-Harris Amendments of 1962, it was often assumed that drugs were safe and effective if they were widely prescribed. Birth defects caused by thalidomide prompted Congressional action to require premarket evidence of effectiveness for the intended use based on “adequate and well-controlled investigations.” 18 Serious consequences related to use of the Dalkon Shield intrauterine device prompted similar legislation for medical devices. 19 Although the merits of prospective evidence collection are now well established, given the accelerating pace of biomedical and engineering discovery, it is imperative that we couple robust evidence standards with an efficient system for evidence generation that can support multiple models of clinical research.
Components of the system: principles of research design
Terminology
As with most endeavors, the first step in driving change is to agree upon terms. The lack of clear, agreed-upon terminology for clinical research contributes to systemic inefficiencies. For example, the terms “pragmatic clinical trial” and “real-world evidence” 20 are commonly used in the lay and expert communities, but both terms are subject to wide variations in definition. This matters because the approach to assuring reliable and high-quality findings and understanding the types of inferences that can be drawn from the studies varies considerably as a function of the specific study design. 21 A foundational element of building a better evidence generation system is reaching consensus on the system we are trying to create and making the relevant description publicly available. To that end, the FDA and the National Institutes of Health have launched a project to develop terminology to enable categorization of clinical research protocols. Other ecosystem stakeholders, including industry, academia, government agencies, and international partners, will be invited to join in this effort.
Quality by design
The Quality by Design framework embraced by the FDA 22 provides a method that, when appropriately adapted to clinical trials, can efficiently align the questions addressed by research and the methods used to answer those questions with the goal of the research. In a clinical trial context, quality by design describes a systems approach in which quality is a key consideration during trial design, with a resultant focus on optimizing trial elements that matter to the credibility of conclusions, avoiding preventable errors, and eschewing processes that are inefficient or detract from the purpose. By defining the elements of trial quality from the start, systems of measurement can be tailored to ensure quality and identify gaps that require reassessment.
For early clinical development and mechanistic research, detailed biological evaluation is essential. For later-phase research designed to inform decisions about which interventions are effective and safe (and often to inform issues of implementation), measurement of meaningful clinical outcomes is likewise necessary. Furthermore, when the goal is to inform health and healthcare decision-making, large numbers of clinical outcomes are often needed to reliably estimate the effects of the intervention. The clinical research enterprise should not be viewed as comprising only two options: either traditional clinical trials, with the plethora of detailed operating procedures that have become accepted as essential to their conduct, or retrospective analyses of clinical practice data. Instead, the system should be viewed as a continuum in which the infrastructure supports multiple types of research. In such a system, the study design is determined by the research question and the assessment of the quality and reliability of the research is based on the design of the study and purpose of the research.
Components of the system: operational infrastructure
The clinical research enterprise
The clinical research enterprise encompasses a broad, complex ecosystem of entities, each with an interest in how research is conducted and which questions are addressed by the research process. Because the results of clinical research findings affect the public, almost everyone has a direct interest. In addition to those organizations typically engaged in clinical research (Table 1), we need a system of evidence generation that also engages research participants and clinicians, particularly those clinicians who are on the front line of clinical care and participants who have been left out of most clinical research because of historical and existing structural biases. As has been the case in our healthcare delivery system, groups including minority populations, elderly persons, those with lower income and lower educational attainment, and rural populations all have been underrepresented in cutting-edge clinical trials.
The Clinical Research Enterprise (CRE): issues and actions

CRE: clinical research enterprise; IRB: institutional review board; FDA: U.S. Food and Drug Administration; PI: principal investigator.
The current clinical research enterprise has a high-quality component that conducts early-phase clinical studies in research clinics that are separated from clinical practice, but this approach is labor-intensive, expensive, and cannot address many critical questions that need to be answered to inform clinical and regulatory practice. Many questions that can only be answered in the context of clinical practice are often left unanswered because of a fragmented approach that disincentivizes collaboration, prompting concerns that efficient research on a large scale is not possible.
Overcoming the challenges of suboptimization
The U.S. healthcare delivery system and the clinical research enterprise as a whole suffer from pervasive suboptimization, a term that refers to the practice of focusing on or incentivizing one component of a total system and making changes intended to optimize the financial and functional activity of that component while ignoring the effects of those changes on the other components. 23 The result is a whole that is less than the sum of its parts. One clear example of suboptimization is the blocking of information. 24 In the business world, possessing information yields a critical competitive advantage. The belief that this is also true in the business of healthcare leads the relevant entities to block information pertinent to risk status and patterns of treatment within health systems—information that, if aggregated, could yield research insights. Much emphasis has been placed on the importance of the flow of information for essential transactional clinical and business issues in healthcare, but there has been less focus on the value of advancing knowledge so that patients are more likely to get the best care over time.
Data infrastructure
Given an appropriate design, successful research is defined by generating data according to a given study protocol and using it to derive inferences that advance knowledge, individual decision-making, or policy. The digitization of science and society is fueling revolutionary opportunities for improving data resources for research. However, these same changes, which have enabled “push-button” data aggregation, real-time interactive measurements, and analysis using prepackaged programs, bring with them the risk of misuse of data for naïve or purposeful reasons. Critical elements of a systematic approach to steering the enterprise to positive outcomes include the development of standard approaches to ensure the quality and integrity of data, integration of different types of data, and enhancement of the ethical and operational construct for data sharing (Table 2). To accelerate these efforts, the education and training of experts in the domains of clinical research, epidemiology, informatics, and quantitative science should be emphasized, as workforce shortages are significant in these areas.
Critical data issues
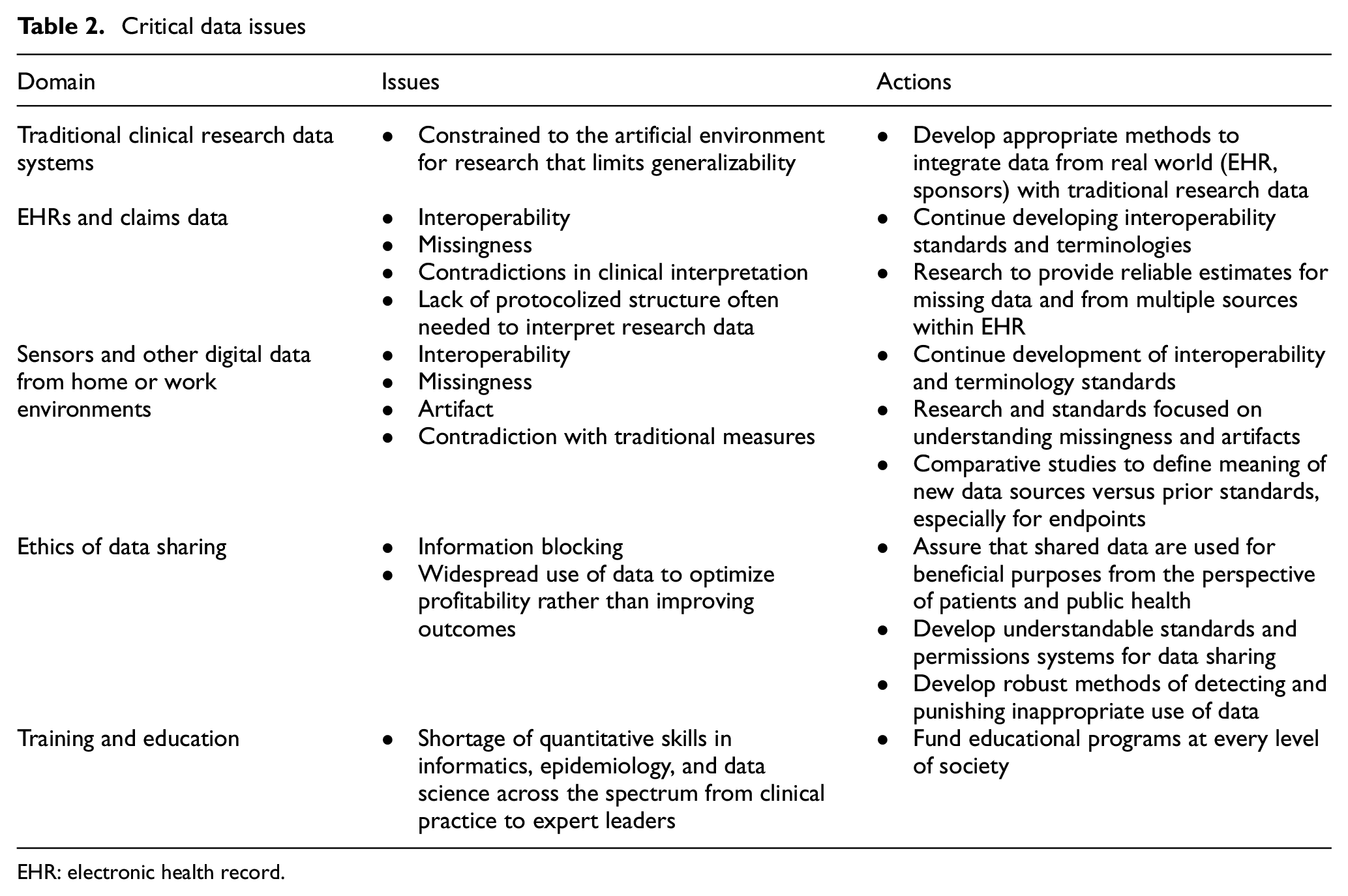
EHR: electronic health record.
Traditional data systems for clinical research continue to evolve and improve with increasing attention to the direct capture of digital information and the use of algorithms to identify discrepancies and errors in real time. Cloud-based data lakes enable the creation of dashboards to inform research operations in real time and to accelerate data cleanup. This part of the clinical research enterprise is doing relatively well with high-quality data that is relevant and can be fit for its purpose, but traditional clinical trials have limitations pointing to the need for other data sources. Traditional clinical trials are expensive and usually involve voluminous data collection and protocols that demand major human resources to ensure compliance. These features often lead to limited sample sizes that may leave open questions regarding performance in clinical practice.
For decades, experts have anticipated the day when electronic health records (EHRs) could form the backbone of the research enterprise. Most Americans now have EHRs, and the quality of the data they contain is now becoming adequate for research. Progress toward “research-ready” elements of EHR systems should deliver more reliable, less expensive data in a variety of clinical settings. The advent of Health Level Seven (HL7) Fast Healthcare Interoperability Resources 25 provides a mechanism for exchanging information among disparate systems, making possible a common data fabric for research. The rate of improvement in EHR quality is accelerating, although key issues such as missing data, data discrepancies, and financial incentives to prioritize the capture of information that optimizes reimbursement (as opposed to care or research) continue to pose challenges. Another critical issue is the incorporation of scheduled assessments that may be required by a research protocol but are not part of routine clinical care.
Just as the long-awaited EHR environment is improving, we are also gaining access to enormous amounts of digital information from sensors and other digital health technologies. These technologies provide a source for real-world physiological measurements gathered outside the healthcare system, including heart rate and rhythm, levels of physical activity, and assessments of cognitive function, among many others.
Until now, most clinical research has been characterized by periodic, intermittent assessments of the research volunteer, done in the artificial environment of the research clinic. But new capabilities for measuring function and symptoms continuously and in routine life settings could enable significant improvements in the degree to which data reflect the actual status of the research participant. The ability to assay symptom status and quality of life using ecological momentary assessment is one example of adding substantial richness to datasets used to evaluate interventions. These tools allow us to improve the relevance of some endpoints to the patient experience, but also have the potential to support more efficient trial designs. As with EHR data, digital information from “real life” presents significant methodological hurdles, including missingness, distinguishing signal from noise, ensuring privacy and security, and developing standards for drawing inferences from complex and novel types of data.
As each of these domains matures, the integration of data and adaptation of regulatory systems, both within federal agencies and across the clinical research enterprise, will present ongoing challenges and opportunities. 26 These different data types may give contradictory measures for a construct such as physical activity or symptom status because the data are acquired in different dimensions of space and time, or because the units of measurement may differ. The regulatory system also faces challenges in dealing with the interpretation of different data sources when new data contradicts or adds different dimensions to a traditionally accepted or validated outcome. For example, the number of steps taken or duration of physical activity may yield different results compared with walking tests done in a clinic.
Uncertainty leads to reluctance to advance the field for understandable reasons. The clinical and scientific worlds have developed cognitive shortcuts that give some data elements primacy, and when different dimensions of data produce contradictory evidence, it can be disconcerting to decision-makers. On the contrary, it is likely that advanced approaches to the use of machine learning and artificial intelligence to derive the best answer from apparently disparate results could lead to better information to support decision-making. In addition, continued innovation in educational programs is needed to produce experts in a much more data-driven healthcare delivery system should lead to a continued acceleration of data quality.
Providing benefits to patients and consumers in a responsible fashion requires serious reconsideration of the ethics, financing, economics, and operational aspects of data sharing. 27 The degree to which the best options for patients and populations can be defined depends directly on the extent to which their data is available for useful purposes. Every person could potentially have access to updated information about the outcomes of “patients like me,” but this will not be possible until we develop robust, societally acceptable approaches to preserving privacy and confidentiality and convince people that sharing data is safe.
This data infrastructure depends on a knowledgeable workforce to implement the many technical improvements that are needed and to guide the system to an outcome that is equitable and effective. Continued innovation in educational programs 28 to produce leaders and experts, but also to prepare the general clinical research and healthcare workforce for a much more data-driven healthcare system will be important.
Operational infrastructure
Ideally, patient groups and clinicians would be aligned on the goal of improving patient outcomes. But despite pervasive rhetoric about “patient-centered care,” financial incentives too often divert focus from the goals of defining effective interventions and implementing them equitably and efficiently. In addition, our EHRs and clinical information systems, despite some improvements, remain optimized for financial applications and not for the generation of needed evidence and support for effective clinical practice. However, progress in this arena is possible, as demonstrated by remarkable improvements in clinical outcomes that have been achieved when disease advocacy groups, including the cystic fibrosis, type 1 diabetes, and multiple myeloma communities, have focused on evidence generation and quality systems in practice. 29
If we can align data systems and regulatory practice with clinical studies designed collaboratively in the interest of patients and their families, the rate and value of evidence generation could grow considerably. Figures 1 and 2 30 show the change from the current fragmented clinical research enterprise to a potential ecosystem configuration of an evidence generation system that could harmonize its many elements to overcome the powerful incentives for suboptimization that currently inhibit collaboration.
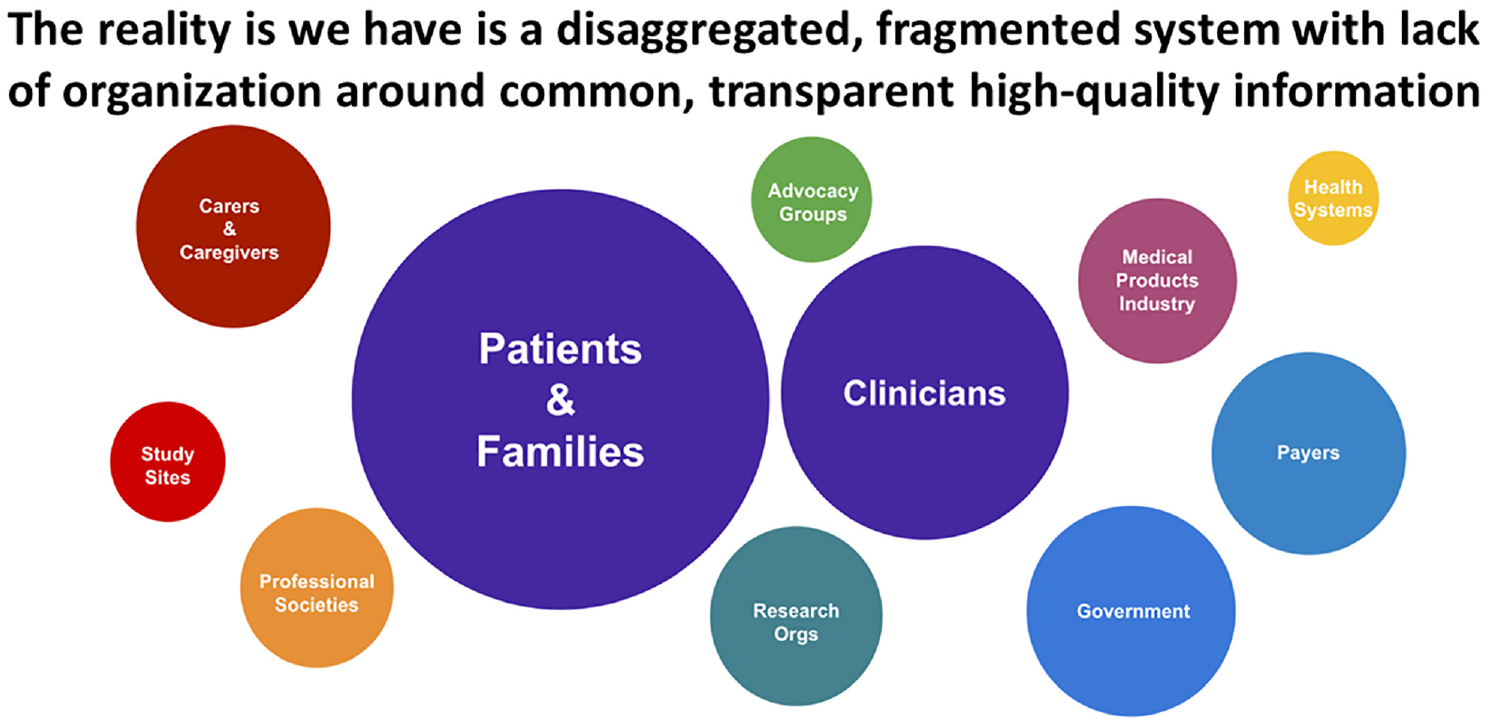
Fragmentation of the healthcare delivery and clinical research systems.
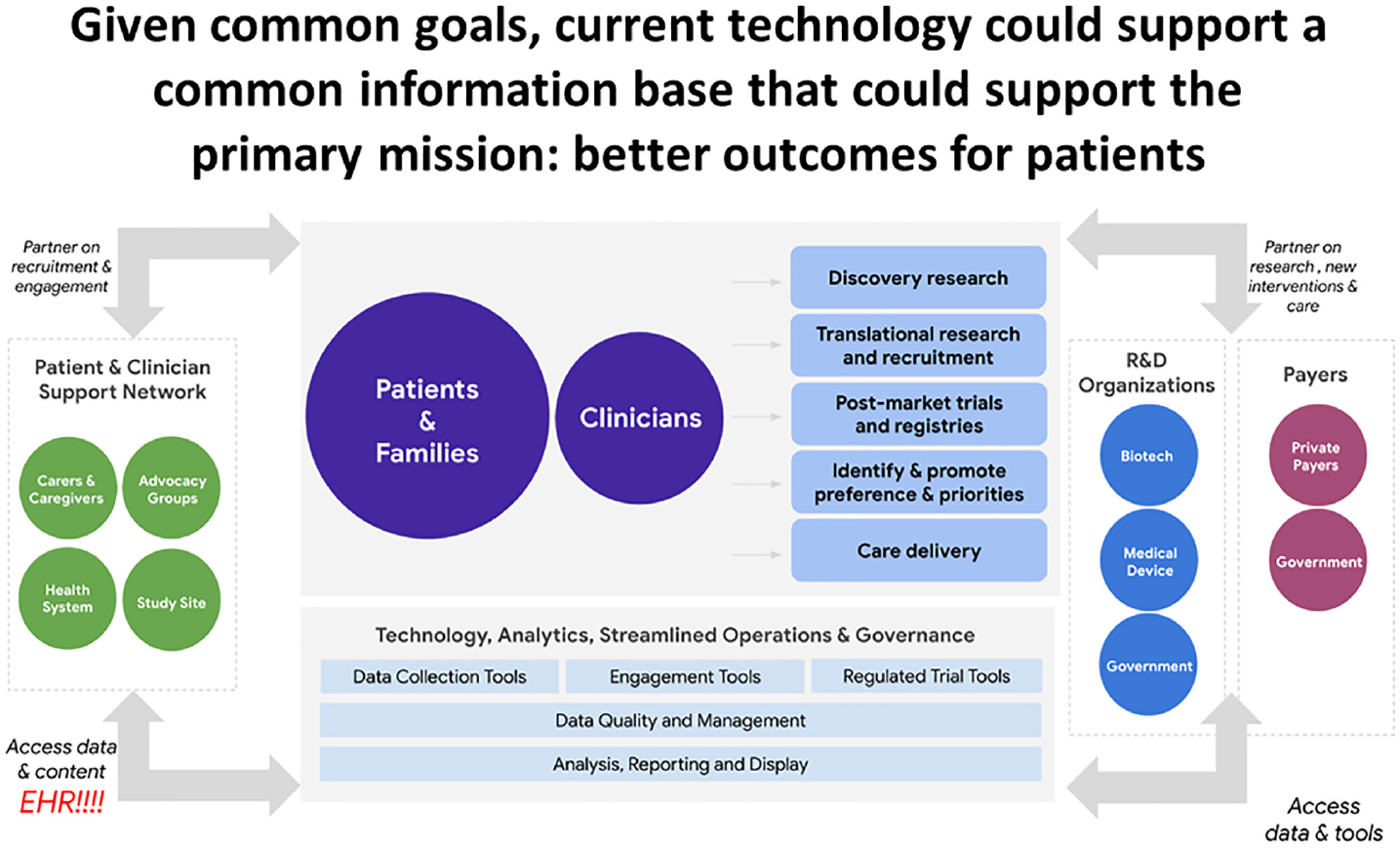
Possible configuration of components and activities needed to overcome systemic fragmentation and create a highly functional healthcare delivery system and clinical research enterprise.
Dissemination and misinformation
The most effective system of evidence generation will benefit from intentionality about dissemination of truthful, reliable knowledge that informs decisions. The details of this issue are beyond the scope of this article, but effective dissemination of reliable information should be a conscious element of an evidence generation system. Although the Internet has enabled remarkable improvements in the dissemination of health information, it has also unleashed profound opportunities to distort or contradict information in a manner that causes people to make bad and uninformed decisions, including decisions that can have a negative impact on their health. 31 This prevalence of distorted information or misinformation is a contributing factor in our decline in health status, including the flattening or downward trend in U.S. life expectancy seen over the past decade, as well as the ground we are losing in fighting common chronic diseases.
The term misinformation describes a spectrum of untruthful or unreliable information that includes disinformation, which is purposefully untruthful information, and malinformation, defined as malicious information intended specifically to harm people. In this new era of misinformation, the promotion of reliable and accurate health information by all components of the clinical research enterprise is an essential feature. The benefits of generating reliable evidence will be attenuated or even negated if that evidence is distorted or misrepresented on a large scale.
Integrating the system: precision health
Given the possibilities afforded by technology, we should be moving toward the goal of assimilating much more high-quality evidence to apply to individual patients in the context of medical care or individual health decisions. This approach, commonly known as precision health, should include the integration of information about the individual and aggregate information about the benefits and risks of possible interventions for different types of patients. For example, the antiviral medication Paxlovid (nirmatrelvir co-packaged with ritonavir) provides benefits for higher-risk individuals, as identified by age or comorbidities, who have contracted COVID. We know this is true because high-quality randomized controlled trials 32 and subsequent real-world evidence 33 clearly demonstrate benefit in high-risk nonhospitalized individuals and minimal benefit to lower-risk individuals. As the quality of evidence about individuals and aggregate information about populations improve, we will enjoy a growing capacity to inform health decisions with comparisons of the expected benefits and risks of those decisions.
The aggregate of this information should also be used to inform health decisions about groups of patients, populations, and the public. 34 Often, broad public health decisions are made with inadequate evidence. However, technological limitations that prevented the collection of needed information have largely been overcome. Particularly for expensive or complex interventions, the optimal decision for the individual may conflict with the best decision for a population because of resource constraints or issues related to deploying interventions. A system of precision public health, however, would enable those decisions to be made explicitly. Integrating these two broad concepts leads to the construct of precision health. By understanding the risks and benefits of interventions at both individual and population levels, we hope to make better decisions at each level.
Conclusion
We now have the technological capacity to move into a new era of evidence generation in which the information needed to inform health decisions, clinical practice, and public health policy occurs in a continuum that is supported by the transparent participation of multiple constituents of the clinical research enterprise. Continued improvements in data science and management, the ethics of data sharing, and alignment of purpose in integrating research and clinical practice are the central areas where focus is needed, with the lattermost element presenting the greatest challenges, particularly regarding the inclusion and involvement of patients and frontline clinicians. Closing the enormous gap between what we need to know for evidence-based practice and what we actually know has the potential to enable major improvements in health status.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.M.C. is an employee of the U.S. Food and Drug Administration. He has no conflicts of interest related to this article to disclose. Prior to his appointment to the U.S. Food and Drug Administration as Commissioner for Food and Drugs, he was an employee of and held equity in Verily Life Sciences and Google Health (Alphabet). He also served on boards of directors for Cytokinetics, Centessa, Clinetic, Keystone Symposia, the Center for Policy Analysis on Trade and Health (CPATH), the Clinical Research Forum, and One Fifteen.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.