
Abstract
Background/Aims:
The US Department of Veterans Affairs Point of Care Clinical Trial Program conducts studies that utilize informatics infrastructure to integrate clinical trial protocols into routine care delivery. The Diuretic Comparison Project compared hydrochlorothiazide to chlorthalidone in reduction of major cardiovascular events in subjects with hypertension. Here we describe the cultural, technical, regulatory, and logistical challenges and solutions that enabled successful implementation of this large pragmatic comparative effectiveness Point of Care clinical trial.
Methods:
Patients were recruited from 72 Veterans Affairs Healthcare Systems using centralized processes for subject identification, obtaining informed consent, data collection, safety monitoring, site communication, and endpoint identification with minimal perturbation of the local clinical care ecosystem. Patients continued to be managed exclusively by their clinical care providers without protocol specified study visits, treatment recommendations, or data collection extraneous to routine care. Centralized study processes were operationalized through the application layer of the electronic health record via a data coordinating center staffed by clinical nurses, data scientists, and statisticians without site-based research coordinators. Study data was collected from the Veterans Affairs electronic health record supplemented by Medicare and National Death Index data.
Results:
The study exceeded its enrolled goal (13,523 subjects) and followed subjects for the 5-year study duration. The key determinant of program success was collaboration between researchers, regulators, clinicians, and administrative staff at the site level to customize study procedures to align with local clinical practice. This flexibility was enabled by designation of the study as minimal risk and determination that clinical care providers were not engaged in research by the Veterans Affairs Central Institutional Review Board. Cultural, regulatory, technical, and logistical problems were identified and solved through iterative collaboration between clinical and research entities. Paramount among these problems was customization of the Veterans Affairs electronic health record and data systems to accommodate study procedures.
Conclusions:
Leveraging clinical care for large-scale clinical trials is feasible but requires a rethinking of traditional clinical trial design (and regulation) to better meet requirements of clinical care ecosystems. Study designs must accommodate site-specific practice variation to reduce the impact on clinical care. A tradeoff thus exists between designing trial processes tailored to expedite local study implementation versus those to produce a more refined response to the research question. The availability of a uniform and flexible electronic health record in the Department of Veterans Affairs played a major role in the success of the trial. Conducting Point of Care research in other healthcare systems without such research-friendly infrastructure presents a more formidable challenge.
Background
Point of Care (POC) clinical trials are an operational approach to clinical research that integrates clinical trial protocols into routine care delivery. 1 These trials leverage clinical resources to improve efficiency, reduce cost, increase patient access to clinical trials, and have potential to enroll patients from community-based clinical settings that do not typically engage in research activities and thereby create study cohorts that are more representative of the disease population. While the approach poses limitations on the questions that can be addressed and the outcomes that can be assessed, selection and creative implementation of trial features into the electronic health record (EHR) enables study designs that range from explanatory trials intended for regulatory submission to pragmatic comparative effectiveness trials for experimental quality improvement as a component of a learning healthcare system. EHRs, often combined with ancillary software, enable centralized recruitment, enrollment, follow-up, data collection, safety monitoring, and outcome ascertainment of study participants and provide for communication between the clinical and research teams.
Engineering requirements for POC trials vary according to trial particulars such as the degree of risk they impose on participants and whether the results are intended for regulatory submission. Beyond these research considerations, studies must be designed with a deep understanding of the incentives and goals for participation by clinical providers and how local clinical workflows can accommodate decentralized activities with minimal perturbation of patient care. In the following sections we briefly describe a POC trial, the Diuretic Comparison Project (DCP), and discuss cultural, regulatory, technical, and logistical considerations of its execution.
Method
The DCP
The DCP is sponsored by the Department of Veteran’s Affairs (VA) as part of the POC Program and has been previously described (clinicaltrials.gov: NCT02185417).2,3 In brief, the DCP is a pragmatic, POC comparative effectiveness trial of two US Food and Drug Administration (FDA) approved diuretics, hydrochlorothiazide and chlorthalidone, on the reduction of major cardiovascular events in patients with hypertension. DCP was conducted at 72 VA Healthcare Systems composed of 72 medical centers and 537 outpatient clinics across the United States and surpassed its target enrollment of 13,500 veterans. The results of DCP are not intended for regulatory review. The VA Central Institutional Review Board (IRB) reviewed and approved the protocol. IRB designation of DCP as a minimal risk study and declaration that clinicians treating study participants were not considered engaged in research had important implications in trial design and budget.
With the assent of treating physicians, who consented to participate in DCP, all veterans over the age of 65 who were prescribed hydrochlorothiazide for hypertension were identified by algorithm from the VA EHR and with the assent of their treating physician were mailed an opt-out introductory letter and informed consent materials (Figure 1). Veterans who had not opted-out by mail were called and those willing to participate were consented and randomized through a centralized process to either continue hydrochlorothiazide 25–50 mg daily or to switch to an equivalent dose of chlorthalidone (12.5–25 mg daily). Participants continued non-protocolized treatment under the supervision of their clinical care providers with no additional study-specific procedures, visits, or data collection. Beyond giving informed consent, assent for each of their patients to participate, and signing diuretic prescription change orders, clinicians had no study responsibilities and importantly, were not considered engaged in research.
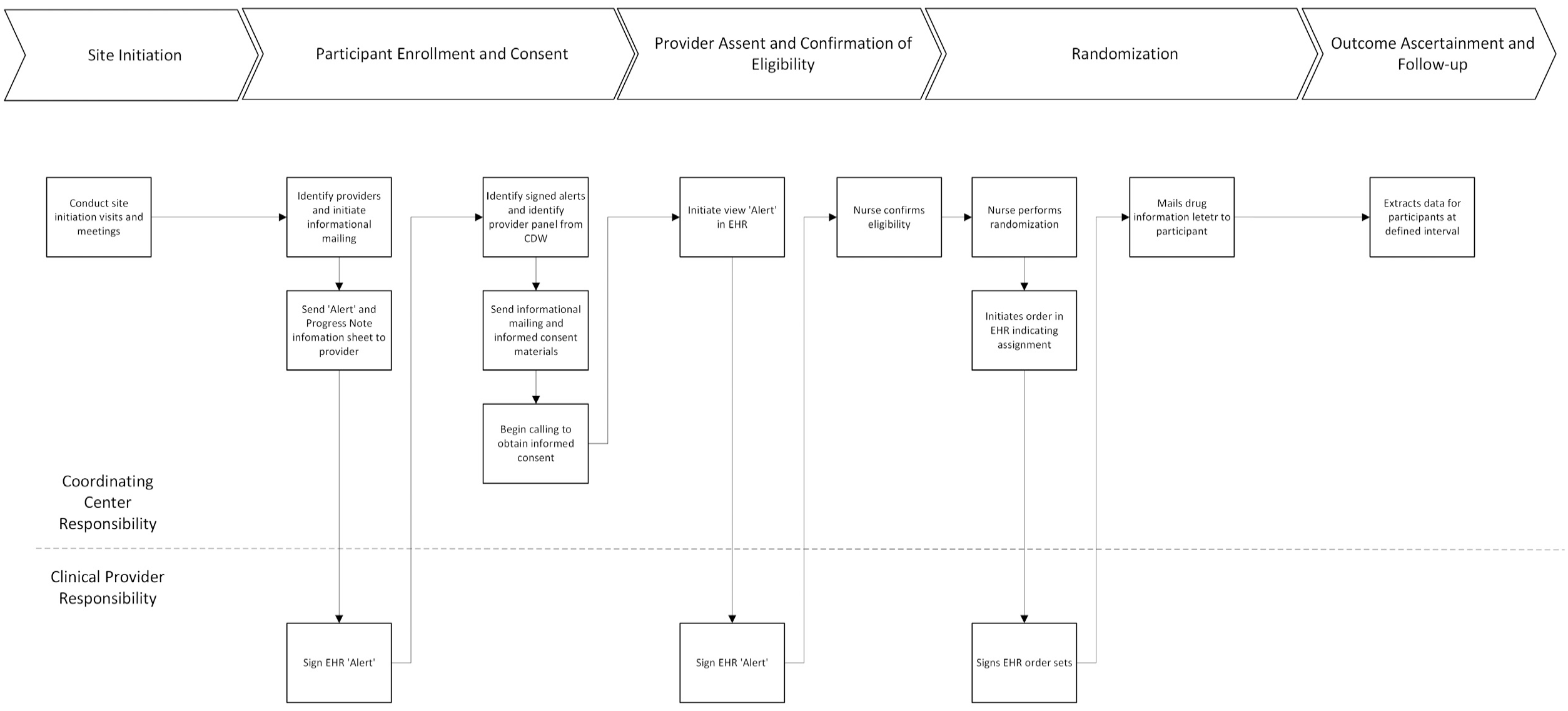
Simplified DCP Workflow.
Results
The baseline characteristics of DCP have been previously published. 3 In brief, 6188 primary care providers were approached for participation in the trial, and 68.7% of them consented to participate. From these 4128 providers, we identified 67,723 patients who were prescribed HCTZ 25 or 50 mg/day and we approached them for participation. Among those eligible, 41,754 had been contacted, 16,595 consented to participate in the study, and 13,523 were randomized. Primary outcomes for 1377 patients were identified from the VA EMR and from the Center for Medicare and Medicaid Services (CMS) databases. In addition, 894 deaths were identified with 3651 incident hospitalizations, and numerous other safety outcomes and adverse events (e.g. abnormal laboratory values, new medication initiations, etc.). Formal reporting and analysis of the results are pending publication at the time of this writing.
Discussion
Cultural considerations of DCP
Most clinical care providers and health care administrators lack a deep understanding of the requirements and processes of clinical research, generally view participation in clinical trials as burdensome distractions, and commonly decline participation. 4 Exceptions include programs that offer a direct and meaningful benefit to clinicians and patients such as access to clinically important therapies that would not otherwise be available (such Precision Oncology Programs).5–8 Studies such as the DCP, where randomization is unlikely to impact importantly on the health outcome of a particular study participant, rely on altruism (from administrators, clinicians, and patients) for clinical integration. Traditional clinical trials intended for licensing purposes supplement altruism with financial incentives, but this is only possible with large study budgets. For DCP and other POC comparative effectiveness studies alternative strategies to engage and incentivize healthcare systems are required.
One such strategy utilized in DCP was to customize research activities at the site-level so that they are highly aligned with clinical workflows and thus present a lower barrier to participation. Prior to launch at each clinical site, we conducted a series of meetings with the medical center leadership and the clinical staff. The meetings set expectations of the research team (such as access to EHR data) and defined, step-by-step, how research activities would be integrated with clinical practice at the site. Importantly, we learned that a “one size fits all” approach to trial processes was not possible and that site-specific modification of operational protocols was required to minimize impact on clinical care. For example, “alert” fatigue, a phenomenon affecting clinical providers who are saturated with electronic clinical reminders and dialogs, 9 was avoided by establishment of streamlined site-specific enrollment processes that minimized clinician clicks in the EHR. While these customizations complicated research activities, they preserved clinician’s autonomy, established trust, and proved vital to obtain local buy-in and reduce time for study rollout.
Interestingly, at some sites buy-in was driven by medical center leadership who saw added value from participation in the study and created incentives for clinician participation. For example, regional directors within the VA allowed a provider’s enrollment of study participants to qualify as a pay-for-performance metric. At sites where this was permitted, both clinical engagement and enrollment were higher than at sites where this alignment of incentives was absent. Enrollment of participants into research programs was important to local leadership as there were national benchmarks for minimal research requirements at VA facilities. The research team further incentivized participation with annual enrollment report cards, and by allowing clinicians access to study data (in a way as to not compromise research integrity) and future opportunity to publish on trial results.
The take-home lesson is that POC studies rely on the engagement of care providers and health care systems as active partners in defining the processes and objectives of the research rather than as passive consumers of its product. Exploiting the full potential of POC methods requires rethinking and redefining traditional cultural and regulatory standards, including incentives for participation, in this research paradigm.
Regulatory considerations of DCP
Enrollment of 13,523 participants was executed through centralized processes without local study staff and follow-up done by non-research clinicians who were not financially reimbursed for their efforts. This was accomplished through invention and iterative refinement of nontraditional study processes created collectively by the research team and the VA Central IRB, Office of Ethics, and Office of Research Oversight with a shared understanding of study requirements, clinical realities, and flexibility of research regulations. The motivation of these groups to reach consensus on non-traditional POC methodology (e.g. reduced safety reporting, non-engagement in research by clinicians, see below) was driven by an understanding (after much discussion) of the limitations of traditional randomized controlled trials (due to cost and time to completion among other factors) to scale and provide answers to many compelling clinical questions.
Two critical IRB determinations enabled successful implementation of this trial. First was designation of the study as minimal risk. Both medications used in DCP have extensive history of use within the VA and in large clinical trials, well-described safety profiles, and were to be administered as per usual care and not through protocolized research procedures. This designation permitted a waiver of documented informed consent requirements and when coupled with a waiver of HIPAA authorization, enabled centralized identification, and telephone-based consent and randomization of study participants with elimination of the requirement to fund research personnel at each site. Finally, the designation permitted reduced safety reporting to the Central IRB that was executed upon continuing review of the trial rather than more burdensome contemporaneous reporting commonly required in traditional or “greater than minimal risk” designated clinical trials. Safety reporting and monitoring by the independent data safety monitoring board was unaltered by the determination and occurred according to standard practices.
The second critical determination was that the participating clinicians were not considered “engaged in research.” Administration of the diuretics was done as part of their clinical care of the research participants rather than as a protocolized research procedure. Rather, the participating clinicians were considered research participants themselves as behavioral data was prospectively collected from them (e.g. they were queried about changes to study medication). This status required informed consent from these participating providers, obtained via progress note templates in the EHR with a waiver of other signed documentation, but eliminated the requirement for research administration and training (e.g. Good Clinical Practice), a major obstacle to clinician participation in POC trials. 4 It also avoided potential conflict-of-interest arising from the dual roles of patient advocate and researcher. 10
Technical considerations of DCP
Execution of a POC clinical trial is critically dependent on integration of study workflows within the application layer of the EHR and data manager access to either its data layer or to a data warehouse that contains frequently updated copies of the EHR dataset. At the launch of DCP the VA Healthcare System utilized a single EHR (computerized patient record system) and provided researchers access to data through the VA Corporate Data Warehouse, a copy of EHR data from the computerized patient record system.
Tools and functionalities resident in the computerized patient record system (application layer) were adapted by Clinical Application Coordinators, highly skilled EHR programmers working at all VA sites, to create study-specific drug order menus and progress notes, clinical alerts, and other forms and workflows to facilitate the trial (Table 1). For example, when patients were randomized to chlorthalidone, standardized order sets were created that discontinued hydrochlorothiazide and initiated an equivalent dose of chlorthalidone. These order sets were created and tested centrally at the coordinating center, “pushed” to each participating site as a computerized patient record system patch and then customized for the local computerized patient record system instance by the Clinical Application Coordinators. Enrolled study participants were then identified in the Corporate Data Warehouse. Importantly, all study activities exposed to the clinical care team were streamlined (requiring as few “clicks” as possible for care providers) and executed through the computerized patient record system, eliminating the need for clinicians to become familiar with a secondary study application, another feature cited as a major obstacle for clinician participation in POC trials. 4
Study process, EHR functions, and supplemental tools used to implement the Diuretic Comparison Project.
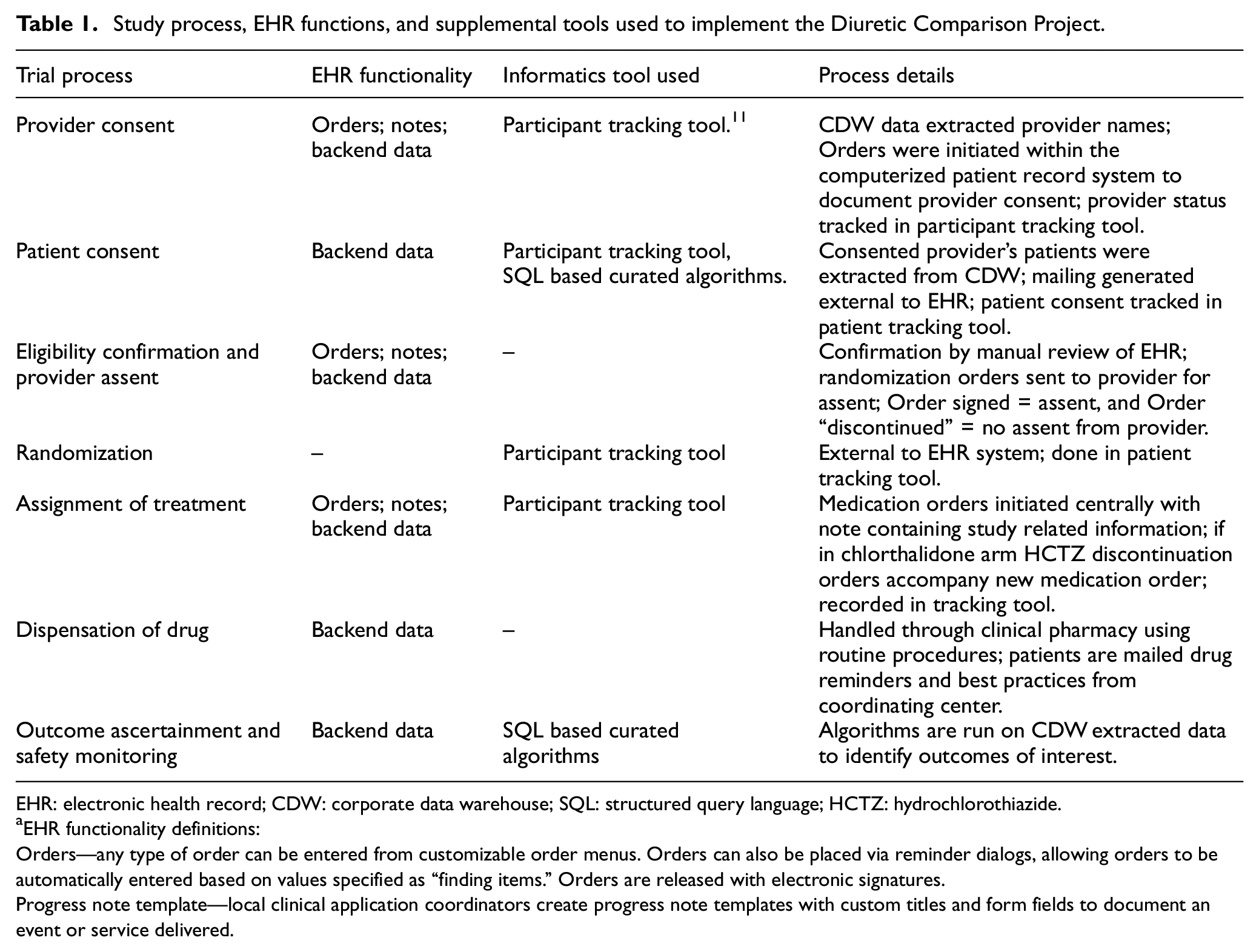
EHR: electronic health record; CDW: corporate data warehouse; SQL: structured query language; HCTZ: hydrochlorothiazide.
EHR functionality definitions:
Orders—any type of order can be entered from customizable order menus. Orders can also be placed via reminder dialogs, allowing orders to be automatically entered based on values specified as “finding items.” Orders are released with electronic signatures.
Progress note template—local clinical application coordinators create progress note templates with custom titles and form fields to document an event or service delivered.
Access to the Corporate Data Warehouse (data layer) by the research team permitted implementation of automated procedures to identify eligible patients, monitor enrolled participants for adverse events of interest, determine time on therapy and vital status, and permit capture of follow-up data at minimal cost. Study outcomes were ascertained from the VA Corporate Data Warehouse using validated complex phenotyping algorithms 12 and from national non-VA datasets (i.e. Medicare data and National Death Index data from the Center for Medicare and Medicaid Services and the Centers for Disease Control, respectively) for study participants who received some care at non-VA facilities. Performance characteristics (sensitivity, accuracy, and positive predictive value) of algorithms used to automate extraction of outcomes through the Corporate Data Warehouse were required to determine that data quality was fit for the study purpose.
The informatics approach described above narrowed the focus of the team to develop, refine, and curate procedures and algorithms to streamline study operations and minimize perturbation of clinical workflows. This highlights that POC studies must be engineered taking full advantage of the flexibility of the EHR application layer to accommodate trial workflows and with near real-time access to the EHR data layer. Absent the ability to customize EHR systems, ancillary software applications that compensate for this deficiency are required with cost, logistical, and behavioral ramifications to trial sponsors.
Finally, a tradeoff exists between designing trial processes tailored to expedite study implementation versus those to produce a more refined response to the research question. For example, introduction of additional (non-EHR derived) data collection would enable interesting sub-analyses and post hoc analyses but introduces additional cost and complexity. Likewise, introduction of a patient reported outcome for this study may have complemented the study outcomes used but would have required workflows and a data capture system not currently available in the EHR. This tension lies at the heart of operationalizing POC trials.
Logistical considerations and implications of DCP
Centralization of all protocol-specified activities relieved clinicians of these responsibilities and reduced the study budget considerably by not requiring staff at sites. Processes performed by coordinating center staff that would typically be done by site coordinators or clinicians included participant identification, recruitment and consenting, drug accounting and management, adverse event detection and reporting, and follow-up study form completion. Participant recruitment and consenting, done via telephone by central callers and not by clinical staff during patient care visits (so-called opportunistic or “hot” recruitment), removed perhaps the most important barrier to clinician participation. 4
Activities that were required of the clinical care team were executed entirely within the application layer of the EHR. First, clinicians gave consent for the Program to screen their patient panel for trial participation. Second, assent of the provider was required to randomize each of their patients who fulfilled eligibility requirements and agreed to participate. Last, the provider was required to continue management of all randomized participants as they would for usual care patients, without reference to a study protocol. This streamlined process minimized the effort required for clinicians to participate in the research and retained their autonomy to determine suitability of the study for individual patients, a major concern of clinicians identified during VA focus groups on POC operational methodology.1,13
POC trials must be engineered to smoothly integrate with the clinical and administrative culture and workflows and EHR capabilities of each participating site. Thus, study protocols cannot be overlayed onto clinical care systems but rather must be designed bottom-up with consideration of the clinical ecosystem(s) in which they will operate. Here we highlight three design decisions that were driven by healthcare system logistics and that had important impacts on the study: restriction of outcome ascertainment to EHR and CMS data, no blinding of treatment assignment and not placing study coordinators at participating sites.
We restricted follow-up data collection to structured elements already resident in the VA Corporate Data Warehouse complimented by linked CMS files to capture care received outside of the VA system. This approach required limiting eligibility to veterans aged greater than 65 years old and participating in Medicare. Because outcomes were defined in DCP using only these available data, they differ from corresponding outcomes from other studies that utilize clinically adjudicated outcomes that make use of confirmatory testing. For example, traditional trials often require measurement of serum troponin to define myocardial infarction as an outcome. This was not possible in DCP as troponin levels could not be ordered by study staff if they were not measured for clinical care and troponin levels are not included in the administrative extracts from CMS. This introduces imprecision in the classification of some outcomes. While this lack of precision is not expected to impact the validity of study results, together with the inability to collect non-EHR outcome data (e.g. Quality Of Life measures), it may make the POC approach to trials less desirable to some study sponsors.
Delays in access to CMS data for research use, often ranging from 18 to 24 months, introduced significant lag times in ascertainment of study outcomes with serious operational impact on the trial including implications for safety review by the independent data and safety monitoring committee. The data and safety monitoring committee made safety and operational decisions often with only half of the expected annual outcomes available. To compensate for this, the committee mandated development and validation of complex algorithms capable of detecting events managed outside of the VA system. This emphasizes the critical importance of understanding data sources and availability when defining outcomes for POC trials and to the extent possible, limiting data sources to those that are readily available and can be monitored for accuracy and completeness. 14
Use of existing clinical pharmacies, that do not have established processes for blinding of medication assignment, mandated that treatment be open label. Had the planning committee determined that blinding was essential, engagement of a research pharmacy, at significant cost and added complexity (due to modification of usual care workflows) would have been required.
Site funding generally constitutes the largest budget item in a clinical trial and is often the major determinant of the feasibility of conducting comparative effectiveness research. Execution of DCP without site funding made the trial feasible from a budgetary perspective but introduced logistical problems. Importantly, site start-up times were prolonged due to delays in approval procedures and modification to the local instance of the computerized patient record system attributed to the lack of designated study personnel to shepherd these activities.
Success of DCP at reaching recruitment goals is testimony to VA commitment to support research activities, executed through a national research and development service and research offices at each clinical facility and by funding a network of VA Medical Centers to facilitate study start-up activities. 15 This infrastructure offset much of the disadvantage of not having study staff on-site. Conducting POC research in other healthcare systems without such research-friendly infrastructure presents a more formidable challenge that requires rethinking of the responsibility of clinical care to support knowledge generation intended to improve the quality of the care they deliver, a concept central to a Learning Healthcare System.
Conclusion
POC trials require study processes that challenge traditional regulatory and ethical tenants of clinical trials and must be developed iteratively through collaboration and compromise between research teams, clinical care systems, and regulatory bodies, with a tradeoff between a refined response to the research question and clinical expediency kept in mind.
POC trials must also meet scientific and regulatory requirements with minimal impact on the clinical ecosystems in which they are implemented. Engineering requirements for these trials vary according to trial particulars, such as the degree of risk they impose on participants and whether the results are intended for regulatory submission. To meet these requirements there is a critical dependence on an EHR that enables integration of study workflows within the application layer and provides access to its data layer, ideally with the capability to modify local study software instances to fit unique clinical environments. Best practices and guidance recommendations for use of EHR-derived data (i.e. “Real World Data”) in POC clinical trials are of paramount importance and are being addressed by academic (e.g. Clinical Trials Transformative Initiative) and regulatory bodies (e.g. Food and Drug Administration).
The most significant hurdle for POC trials is attaining and maintaining site participation, particularly when altruism is the primary motivator. Minimizing study responsibilities for clinicians (e.g. through centralization of study processes) is essential. Central IRB designation of DCP as a minimal risk study and declaration that clinicians treating study patients were not considered engaged in research had important implications in this regard, particularly with respect to relieving clinicians of the administrative overhead associated with research participation (i.e. responsibility for Good Clinical Practice training, IRB review submissions, safety reporting).
Implementation of POC methodology at scale, particularly to address comparative effectiveness questions relevant to healthcare delivery, requires support and infrastructure from clinical care services that must come to view active participation in knowledge generation as a necessary component of decision support and learning activities within a healthcare system. A critical advantage (and thus incentive) for sites participating in POC trials is that study results are “locally selfish” and can be directly implemented in decision support tools, eliminating the “T2 Translation Gap.” Acceptance of results of POC trials by other healthcare systems, that did not participate in the research, is accompanied by the usual concerns of generalizability that pertain to traditional clinical trials.
Continued development of POC methodology requires a codified framework to approach ethical, cultural, technical, and regulatory aspects of this research.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by the Cooperative Studies Program of the Department of Veterans Affairs Office of Research and Development. The opinions expressed in this article are those of the authors and do not necessarily represent those of the Department of Veterans Affairs or United States Government. No disclosures to note for this study.
Trial registration
ClinicalTrials.gov Identifier: NCT02185417