
Abstract
Advancement in science & technology of medical industry is coming up with innovations. Medical devices play a vital role. Real Time Location System (RTLS) which includes Radio Frequency Identification (RFID) device, Wi-Fi technology that helps to look after adverse events and recall products when required, thereby, prevent theft by keeping regular check on their movement with location sensors. In 19th century two products of United States faced product withdrawal. After that, US felt the need to develop system for product record management and tracking. In 2004, FDA introduced Unique Device Identifier (UDI) for medical device tracking. With time, GHTF came up with UDI working groups and in 2013, new version of UDI guidelines for globally harmonized use was generated. In this article study about various countries has been done. It was found that countries like United States and Europe have introduced medical device tracking system and by 2022 this will be implemented in India.
Keywords
Introduction
Technology is reaching new heights with time. Due to innovation in various fields. Man has become accessible to various things through computerized technology in a short time. The Healthcare System is also showing innovations in medical equipment.
1 Electronic Health Records (EHR) advent of the electronic system helps largely record medical and surgical equipment as improved diagnosis and treatment to various diseases using medical devices. Before the tracking of medical devices came into existence, it was difficult to keep an eye upon the journey they went through from their manufacturers to their disposal. With passage of time, Real Time Location System (RTLS) comes into light making it convenient to access movement of products and recall products with errors in their functioning or showing adverse effects. Examples of RTLS are Radio Frequency Identification Device (RFID), Wi-Fi system, Bluetooth, cellular data etc. 2 Unique Device Identification (UDI) system is the latest tracking system used for medical devices. It is available in Automatic Identification and Data Capture (AIDC) and Human Readable Interpretation (HRI) formats on various levels of packing or labels of products. Each product is provided with a unique numeric or alphanumeric combination code. 3 IMDRF is looking forward to mandating UDI system in the coming future for global harmonization to aid healthcare system to track medical devices and collect patient information in less time. 4
Medical device tracking techniques
There are various techniques used for medical device tracking. RTLS has made it convenient to access movement of products with help of RFID, Wi-Fi system along with other technologies like Bluetooth, cellular data etc. 5
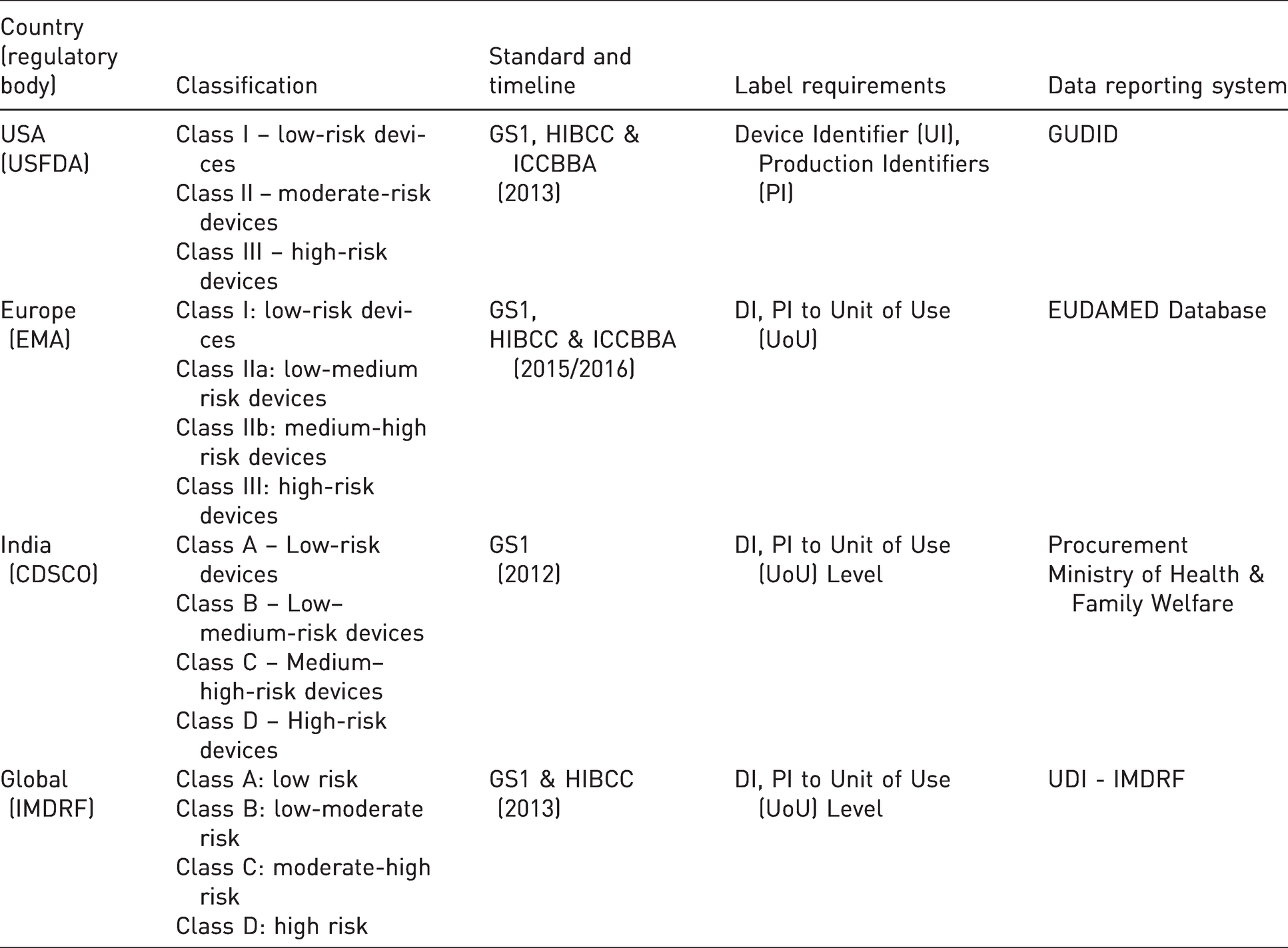
In med-20th century RFID technique was developed and commercialized in 1970. It evolved with the benefit to improving patient care quality by tracking products and people's information. RFID uses an interrogator, working on radio waves to locate and identify tags to read. 6 UDI is the latest globally accepted tracking system for medical devices with Health Industry Business Communications Council (HIBCC), Global Standards One (GS1), International Council for Commonality in Blood Banking Automation (ICCBBA) standards on product labels and package, in AIDC and HRI formats.7–9
Global regulatory scenario
Development of medical device regulations is an important aspect to gain public faith and confidence. In some countries, medical device tracking is done. However, there are many countries with no device tracking regulation. The origin of medical device tracking regulation in the US, EU and India is shown in Figure 1. Such countries should regulate tracking by licensed government-approved authorities to aid authentic reliable products to reduce counterfeited and flawed devices in the market. International Medical Device Regulators Forum (IMDRF) have issued UDI guidelines for harmonized medical device tracking across the globe, to ensure safety, efficacy and quality to lift public health by providing the genuine product. 15
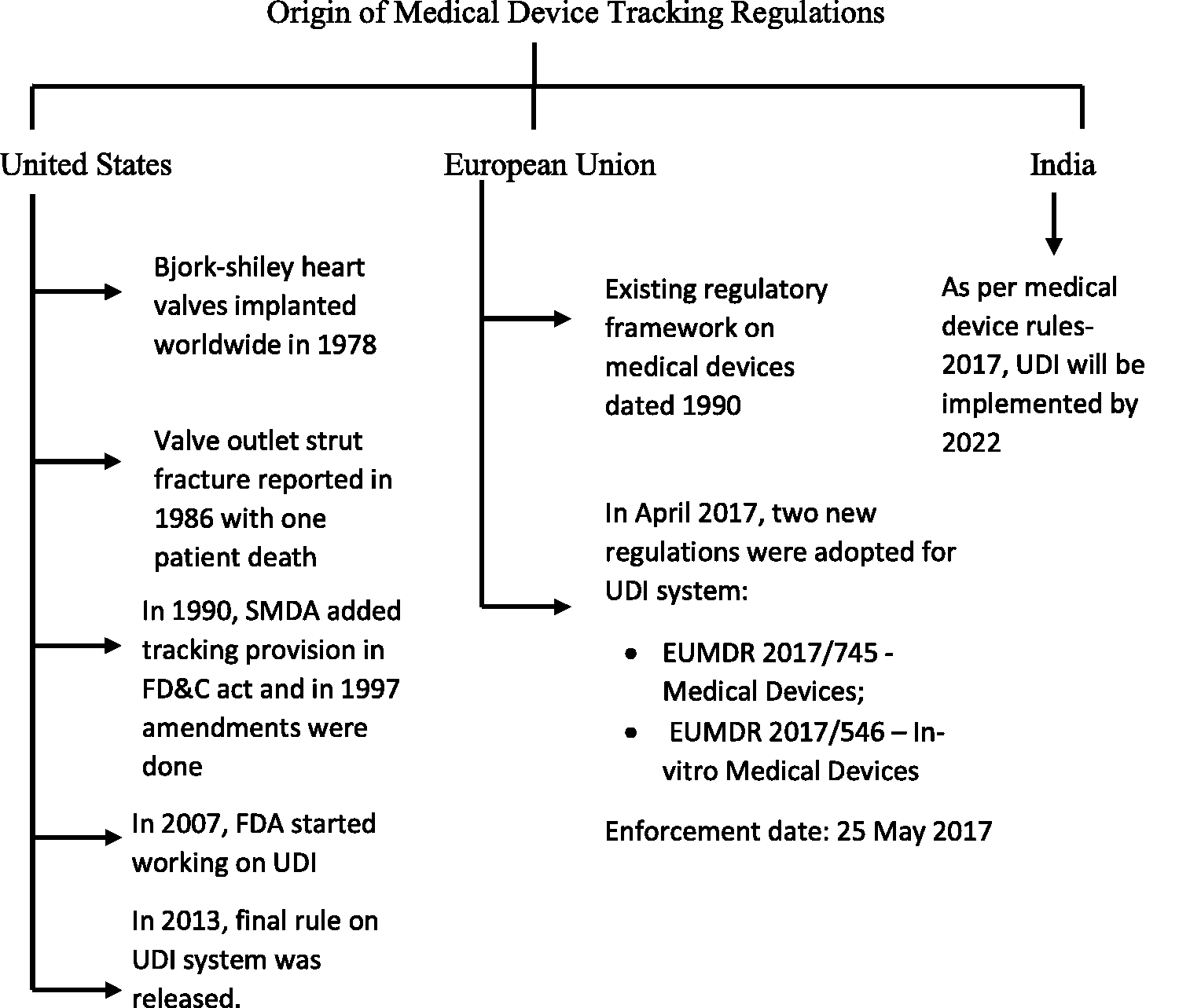
Harmonized tracking systems of medical and surgical devices will ensure easier identification and tracking of products on the supply chain. It will also reduce cost and time for marketing medical devices accompanying strengthened public confidence on marketed products.16–18
United States
Origin of device tracking
Bjork-Shiley heart valves were implanted worldwide since 1978. Valve outlet strut fracture (OSF) was reported in 1986, which resulted in one patient’s death, making it a potentially life-threatening risk. Complete serial numbers of 97% valves were identified from the manufacturer’s shipment and hospital records. Still, clinical records for 16% of valves were missing due to improper implantation centers record storage policy. During this case, the concept of tracking came to light. Manufacturer did not have any means to contact patients and physicians like the tracking system that exists today. Later, requirement of a system for maintenance of records was passed by US congress legislation. Ventritex invoked the use of tracking system to keep the patient’s information. 10
In 1994 and 1995, implantable cardioverter-defibrillator (ICDs) Ventritex cadence models V-110 & V-112 respectively received approval from the Food and Drug Administration (FDA). These devices were used to protect patients from tachyarrhythmia. In 1997, a patient death due to device malfunctioning was reported to manufacturer. After the analysis of the device potential reason for device failure was noted. As a result, device software was modified to minimize device adverse effects. The FDA immediately approved Ventritex for device notification and new software version. Manufacturers began device notification by contacting physicians. They also tried to contact patients by sending certified letter at their address. Ventritex used device tracking system to maintain the device and patient information.
The Bjork-Shiley Convexo-Concave heart valve and the Vitek temporomandibular joint implant faced product withdrawals before the implementation of the device tracking regulation. 11
In 1997, The Modernization act was enacted giving freedom to the FDA to decide which device should be a part of the medical device tracking program. 19
Earlier tracking regulations were implemented for certain devices in US. In 2004, FDA came up with innovative invention of UDI. UDI assist healthcare workers and patients, manufacturers to withdraw medical devices with defects/serious adverse events, from the market. It also helped to inform patients and healthcare professionals regarding the same. Regulations & guidelines for medical device tracking are found in 21 CFR Part 821 and “Medical device Tracking Guidance for Industry and Food and Drug Administration Staff”. A medical device must be used as per Standard Operating Procedure (SOP) provided by the manufacturer. End distributors should provide patients information to the manufacturers.20,21
Manufacturers are required to track and recall certain products having adverse risks to health on the orders of the agency under The Food and Drug Administration Modernization Act (FDAMA). In 1990, under the Safe Medical Devices Act (SMDA) tracking provision of sec. 519(e) of the FD & C act was included. 12 Timeline for UDI compliance in the United states is shown in Table 1.
Timeline for compliance with UDI requirements. 22

Unique device identifier
UDI codes are unique for each medical device. They are formed with a combination of numeric or alphanumeric characters. They are present on outer & inner packing of products and on labels with some exceptions for certain devices. It should be present AIDC and HRI formats. 23 It focuses on improving post-marketing surveillance along with patients’ safety facilitating medical devices innovation.
UDI-DI (Device Identifier) & UDI-PI (Product Identifier) collectively form UDI. UDI-DI portion contains information related to the device model/version. UDI-PI portion is not fixed or specified by the labeler it contains lot/batch no.; serial no.; expiration & manufacturing date of specific device. It's mandatory to have UDI-DI but the inclusion of UDI-PI depends on conditions.24–26
GUDID (Global Unique Device Identification Database)
FDA administered GUDID is used to obtain device-related information, it contains only the UDI-DI part of UDI information submitted by the device manufacturer. 27 FDA issued guidelines on GUDID for industries on 24th June 2014 “Global Unique Device Identification Database (GUDID): Guidance for Industry” describing key concepts.28,29
Benefits
Helps to correct the defects and problems in device by accurate identification
Reduce health consequences due to medical errors.
Strong post-market surveillance system
Device analysis
Secure distribution chain
Effective product recalls 30
21cfr Part 821 tracking requirements
FDA has defined certain criteria for (class II & III) devices tracking. Devices meeting the criteria and falling under section 519(e)(1) must be tracked as per the orders of FDA. Manufacturer must provide the following information to FDA about devices:
○ Except as required, under Section 518(e), before distribution of the tracked device to patient information about distributors must be provided, upon FDA request within 3 working days. ○ For a lifelong single patient use device, the following information should be provided to FDA within 10 working days upon request: i The essential information for tracking the device ii Device shipment date iii Patient details with patient device receiving date iv Physicians detail monitoring patient with device v Details of patient and physician involved in device explanation, patient death date; or device returning date. ○ Expect as per section 518(e) of the act, multiple patients use devices information about device, device tracking, distributor, patient and physician should be provided to FDA within 10 working days upon request.
A manufacturer must provide SOP for auditing, collection and data management. It should also provide device tracking information to FDA upon request.31–34
Warning letter
In 2010, the FDA inspected Philips Medical Systems (Seattle, Washington). They found their devices were adulterated and not manufactured as per Current Good Manufacturing Practice (CGMP) requirements of the Quality System (QS) regulation found at Title 21, Code of Federal Regulations (CFR), Part 820. Other violations were also found including failure to establish and maintain adequate procedures for device tracking as required by 21 CFR 821.25(c). 35
IMDRF (formerly GHTF)
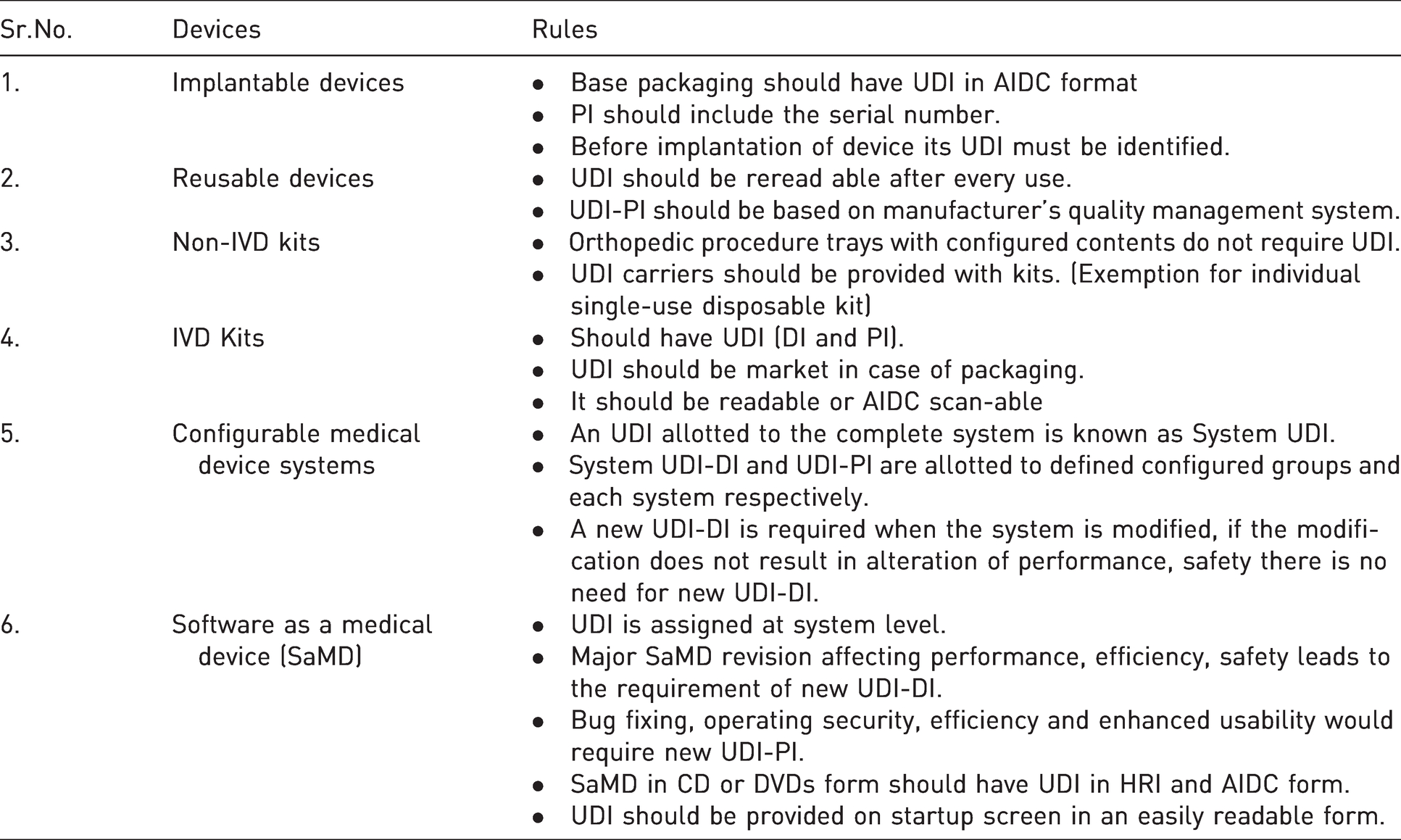
In 2008, GHTF provided global regulatory authorities with UDI working group. On 9 December 2013 IMDRF set in new version of UDI guidelines. These guidelines were formulated for globally harmonized approached to track medical devices with an identical framework of UDI system. It aims towards identifying medical errors, adverse events and fundamental device data capturing to increase patient safety and care by analysis of problem and recall product when necessary. 36 Rules for specific device about UDI is shown in Table 2.
Rules for specific devices. 37
•
○ UDI Carrier
○ Medical Device UDI should be globally readable
○ UDI should not be regionally modified/standard UDI
○ Database should be used for data submission
○ All medical Devices should have UDI (expect exemption)
•
○ UDI Carrier application
○ UDI creation and maintenance
○ UDI Database (UDID) for data elements
○ Establishment of common database for collection and exchange of data in web-based interface HL7 SPL formats.
•
Each device should have its unique UDI.
○
○ Medical Devices are earmarked/designed with UDI-DI at the level unit of use (UoU) when UDI is not provided to an individual medical device
○ Kits should have UDI.
○ Any change in device information needs new UDI-DI. Minimum changes should be updated in database within 30days of change made to prevent difficulty in device tracking.
•
○ It should be in AIDC and HRI format
○ For class A and B one-time useable medical devices, UDI should be on higher/outer package of product.
○ In case, of non-prescription medical devices, UDI is not required in AIDC encoded form.
○ In case of limited space, AIDC format should be preferred.
○ For reusable devices, UDI should be on device itself, so that it can be reprocessed after every use.
•
○ The use of UDI core elements should be supported.
○ Labeler should not include any confidential information in UDID.
○ The manufacturer is responsible for initial submission and updates about device information in UDID.
○ An appropriate method for data validation should be implemented.
○ The manufacturer shall regularly verify devices related data.
○ Core element data should be accessible to the public free of cost.
○ The submission of UDI-DI in UDID does not confirm its authorization to jurisdictions.
○ Any change should be updated within 30 days.
○ HL7 SPL should be used for data submission and update.
Europe
In April 2017, the European Union introduced two new regulations for Medical devices and In-vitro medical devices on UDI system.
EUMDR 2017/745 – Medical Devices EUMDR 2017/546 – In-vitro Medical Devices
The UDI system was introduced for easy monitoring and tracing of medical devices. It helped to detect medical errors, falsified device, for better post-marketing surveillance of medical devices (exception for custom-made and investigational devices). 13
According to Article 27 & 24 of medical devices and In-vitro medical devices respectively, UDI consist of the following:
○ UDI-DI & UDI-PI ○ Must be placed on label of medical device ○ Must prepare the UDI database for data collection as part of EUDAMED database.
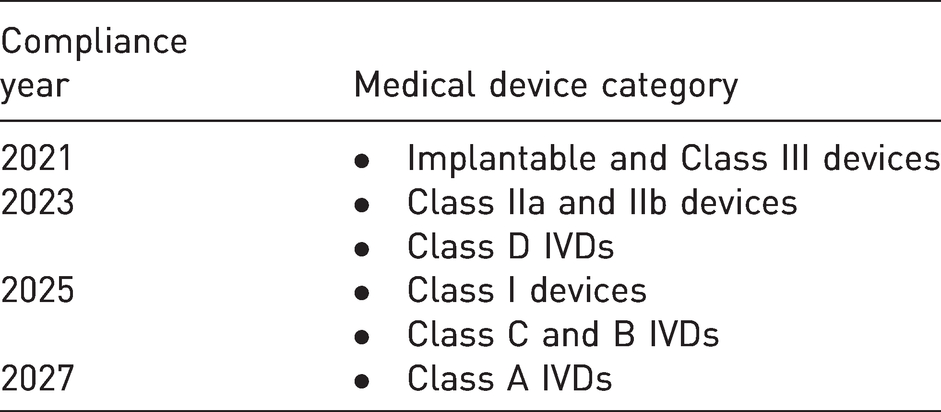
UDI is a unique code for each device, it must be present on a device and its higher packaging (excluding outer packaging). In case of reusable devices, they should be placed on device itself. Manufacturer must make sure that correct information is uploaded to UDI database. 39 Timeline for UDI compliance in EU is shown in Table 3.
Timeline for compliance with UDI 40 .
UDI registration rule
○ According to rules in article 27(2), manufacturer must assign a unique UDI-DI to a device (except custom-made device). Information should be uploaded to UDI database along with other essential information.
○ As per rules of Article 22(1) and (3), before placing a system or procedure pack in market manufacturer must provide them with UDI-DI and upload it with UDI database.
○ In reference to Article 52(3) and Article 52(4), devices subjected to conformity assessment must be assigned with basic UDI-DI.
Manufacturer must enter and verify information. It should be kept up to date as per Annex VI Section 2(A) for medical devices except custom made devices in market. 41
Timeline for compliance with UDI requirements
As per EUMDR 2017/745 and 2017/746 after the implementation of UDI system regulations, deadlines for assigning and registration of UDI with EUDAMED have been provided by regulatory authorities, ensuring manufacturers failing to comply with these regulations would not be allowed to supply items in EU.
Placement of UDI on the package or label of device
○ UDI can be in the form of AIDC or HRI technology/both on the label/package of device (in case to limited space on label).
○ In case of limited space then AIDC should be preferred.
○ HRI format should be used for home care devices.
○ For single-use devices, UDI must be present on higher level of packaging. 40
Requirement of a new UDI-DI
Any change in product information like device name, model number, labeling changes, warnings, quantity variation, etc. and problem in tracking and identification of medical device new UDI-DI is required. For changes that don’t require new UDI-DI, manufacturer must update database within 30days before making changes effective. 41
India
In the pre-COVID-19 era demand of medical devices were less, COVID-19 crises have boosted sale of various healthcare products and medical devices. This has increased the number of unauthorized sellers in the supply chain selling spurious products. In the absence of specific guidelines for tracking of medical devices, many countries succeeded in exporting low-quality products in our country.
Various protective equipment has been imported from China. Over 60,000 PPE kits and masks have been imported and distributed to various health care workers in India. During the quality check, PPE kits doesn’t meet the quality criteria and failed the test. Without any proper regulation for tracking, it is made difficult to inform users about the low quality of protective equipment to prevent health issues. 42
There are many products in the market with improper labeling. GS1 has introduced a “Smart Consumer” application for customers. This application has provided access to product information by scanning a barcode and making it easy for a consumer to differentiate between different products. This app. has also helped in tracking and checking the availability of the product in the supply chain. 43
cTill date, there is no strict regulation and guidelines by the Government of India (GoI) to track medical devices concerning patient safety, life sustenance and medical device theft. Drug Technical Advisory Board (DTAB) recommended formation of tracking system concerning patient safety, due to poor tracking system, in Johnson & Johnson hip implant case government was unable to track maximum number of patients with faulty hip implants. 44
In 2010, Central Drugs Standard Control Organization (CDSCO) and Pharmacy Council of India (PCI) formed Pharmacovigilance Programme of India (PvPI) for serious adverse events (SAEs). By 2015 Drug Controller General of India (DCGI) introduced Materiovigilance programme of India (MvPI) for monitoring adverse events related to medical devices. It created awareness among patients and healthcare professionals about benefits and harmful effects asociated with falsified, defected medical devices. 14
Currently, lot no., batch no., assigned by the manufacturer to medical devices are used to track their movement. Manufacturers must keep a record of adverse events, complaints received by healthcare professionals related to medical devices.45,46
As per UDI & Medical Device Rules-2017, chapter VII, rule 47 UDI system will be effective from 2022. According to the new policy, all manufacturers and importers must comply according to them. 47
UDI emergence will benefit manufacturers, patients and healthcare system by enabling:
○ discovery of falsified medical device with faster recall process, ○ reduced medical errors, ○ better assessment of device performance, ○ reduced counterfeiting etc.
Future perspectives
Expanding technology in healthcare systems is contributing to increased health benefits. With better identification, recalls of falsified medical devices, reduced medical errors, decreased counterfeiting, improved device assessments, to improved public health. UDI system is serving as an asset in the collection, saving, and extraction of medical device information for its safe and effective use for identification and tracking. In the coming future, UDI is aimed to be mandated globally with a harmonized regulation system for optimized use. Though there will be challenges in the beginning for manufactures to comply with new regulations that would affect their cost and system. However, with regular use, its benefits will overweight its appliance value.14,45
Comparison
The comparison between United States, Europe, India and global (IMDRF) regulations is shown under the Table 4.
Conclusion
Modernization and progressive technology are reaching new height with passing time. They are becoming more efficient and effective with new ideas to do something innovative and deal with problems. Healthcare department is also pushing boundaries of technology to get better treatment and diagnostic methods. Tracking of medical devices is also becoming an important part of medical devices industry for safety of patients, become possible with RTLS.
Unique Device Identification system is the latest technology used to track product movement and collect patient information. It has shown positive consequences in providing authentic products, increase patient safety, reduce product recall time. UDI implementation turns up to be costly at initial stage but its benefits weigh its cost.
IMDRF has formulated framework on UDI for regulatory authorities intended to develop UDI system. Different countries are following their own set guidelines on UDI that won’t help to achieve globally harmonized system of UDI, these differences should be eliminated. In coming era, UDI will be implemented globally at every level of packaging. In UDI database management any small modification in medical device requires data alteration in database, sometimes new UDI-DI is also required to overcome this issue. advancement in technology is required for data management more precisely.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.