
Abstract
Metformin hydrochloride remains the cornerstone oral therapy for the treatment of type 2 diabetes mellitus (T2DM), with millions of patients relying on its sustained glycemic control and safety profile. Classified under the Biopharmaceutics Classification System (BCS) as a Class III compound—exhibiting high solubility and low permeability—it qualifies for regulatory biowaivers under stringent in-vitro dissolution criteria. These regulatory waivers eliminate the need for in-vivo bioequivalence studies, significantly accelerating the approval of generic formulations, particularly in resource-limited settings. This study evaluates and compares the in-vitro dissolution profiles of five commercially available metformin hydrochloride 500 mg tablet formulations under simulated gastrointestinal conditions. Dissolution testing was conducted using USP Apparatus II (paddle method) across three media—0.1 N HCl (pH 1.2), acetate buffer (pH 4.5), and phosphate buffer (pH 6.8)—to mimic stomach and intestinal environments. The dissolution profiles were assessed using similarity factor (f2), difference factor (f1), and dissolution efficiency (DE) as recommended by regulatory authorities. The findings reveal that while generic brands show comparable dissolution to the innovator product in selected media. In this study, almost all five formulations released more than 85% of the drug within 15 min under the three tested conditions, suggesting compliance with the “very rapidly dissolving” criterion set forth by regulatory authorities. However, variations in f1, f2, and dissolution efficiency metrics among brands indicate differences in formulation technology, excipient selection, and manufacturing consistency. This study affirms the value of robust in-vitro dissolution testing as a surrogate for bioequivalence, emphasizing its importance in regulatory science, generic drug development, and public health policy. Our results call for regulatory vigilance and formulation refinement to ensure therapeutic equivalence and support global diabetes management initiatives.
Introduction
Background
Diabetes mellitus, particularly type 2 diabetes mellitus (T2DM), represents one of the most significant public health challenges of the 21st century.1,2 According to the International Diabetes Federation (IDF), over 537 million adults are living with diabetes globally as of 2023, a figure projected to rise to 643 million by 2030. 3 The economic burden is equally alarming, with global healthcare expenditures for diabetes surpassing USD 966 billion in 2021 alone. 4
Metformin hydrochloride is universally recommended as the first-line pharmacotherapy for T2DM. It functions primarily by reducing hepatic glucose production, enhancing insulin sensitivity, and improving peripheral glucose uptake. As a result of its safety, efficacy, and affordability, metformin is included on the World Health Organization’s Model List of Essential Medicines.5,6
Biopharmaceutics Classification System (BCS) and regulatory relevance
The Biopharmaceutics Classification System (BCS) is a scientific framework developed to classify drugs based on their aqueous solubility and intestinal permeability. It serves as the basis for granting biowaivers—regulatory approval without in-vivo bioequivalence studies—based on in-vitro dissolution testing.7,8
Metformin hydrochloride falls into BCS Class III, which comprises highly soluble but poorly permeable drugs. According to the FDA, a drug substance is considered “highly soluble” when the highest single therapeutic dose dissolves in 250 mL or less of aqueous media over the pH range of 1–7.5 at 37°C. Although low permeability typically necessitates caution in interchangeability, the rapid dissolution of BCS Class III drugs allows for biowaivers when formulations exhibit similar disintegration and dissolution across media.9,10
The WHO, US FDA, and European Medicines Agency (EMA) has endorsed BCS-based biowaivers for Class III drugs under specific conditions. These include use of immediate-release formulations, highly similar excipient compositions, rapid dissolution (≥85% within 15 min) in all three media and Consistency across batches.11–13
Importance of in-vitro dissolution testing
Dissolution testing provides vital insights into the rate and extent of drug release from a dosage form. It acts as a surrogate for in-vivo bioavailability and plays a pivotal role in formulation development, quality control, and regulatory submission. For BCS Class III drugs, dissolution testing is even more critical due to the reliance on in-vitro parameters to establish therapeutic equivalence.14–16
The similarity factor (f2) and difference factor (f1) are commonly used statistical tools to compare dissolution profiles. An f2 value >50 indicates profile similarity, while an f1 <15 denotes acceptable differences.17,18 Dissolution Efficiency (DE) is another valuable parameter representing the area under the dissolution curve as a percentage of the total area, facilitating more nuanced comparisons.19–22
Market and public health implications
The availability of affordable and therapeutically equivalent generics is essential for health equity, especially in low- and middle-income countries (LMICs), where branded medications are often unaffordable. 23 However, therapeutic failures and adverse drug reactions may arise if generics are not pharmaceutically equivalent to innovator products.23,24
Given that metformin is often used in long-term, sometimes lifelong, treatment regimens, any deviation in its release profile can significantly affect glycemic control and increase the risk of complications like cardiovascular disease, nephropathy, and retinopathy. 25
Aim of the study
This study aims to Evaluate and compare the in-vitro dissolution profiles of five metformin hydrochloride 500 mg tablet formulations, to Determine their suitability for biowaiver approval based on regulatory criteria and to Highlight the need for ongoing quality assurance and regulatory oversight. The findings will provide evidence for manufacturers, regulators, clinicians, and policymakers to ensure that patients receive safe, effective, and interchangeable metformin formulations.
Materials and methods
Materials
This study included five different commercially available brands of metformin hydrochloride immediate-release tablets, each containing 500 mg of the active pharmaceutical ingredient (API). Brands were selected based on market availability, prescription frequency, and geographic distribution. Only tablets labeled as immediate-release and intended for oral administration were considered.
Metformin hydrochloride reference standard
Hydrochloric acid (HCl) – Analytical reagent grade
Acetic acid and sodium acetate – For acetate buffer (pH 4.5)
Potassium dihydrogen phosphate (KH2PO4) and sodium hydroxide (NaOH) – For phosphate buffer (pH 6.8)
Deionized water – Used in all solution preparations and dilutions
Whatman filter paper (Grade 1) – Used for sample filtration.
All chemicals and reagents were of analytical grade and used without further purification.
Instrumentation
To ensure accurate and reproducible results, validated instruments were employed throughout the experiment: Dissolution Apparatus – USP Apparatus II (Paddle Method, Electrolab TDT-08L) UV-Visible Spectrophotometer – Shimadzu UV-1800 with quartz cuvettes (1 cm path length) pH Meter – Sistronics, calibrated with standard buffer solutions (pH 4.0, 7.0, and 10.0) Analytical Balance – Sartorius, calibrated for 0.1 mg sensitivity Water Bath – Thermostatically controlled (37 ± 0.5°C) Magnetic stirrer – For buffer preparation and degassing Sonicator – For degassing dissolution media
Dissolution media preparation
Three dissolution media were prepared according to regulatory guidance: N HCl (pH 1.2): Prepared by diluting concentrated hydrochloric acid in deionized water to simulate gastric conditions. Acetate Buffer (pH 4.5): Prepared using acetic acid and sodium acetate in deionized water, adjusted to the required pH. Phosphate Buffer (pH 6.8): Prepared by mixing KH2PO4 and NaOH and adjusting pH to simulate intestinal conditions. All media were freshly prepared, degassed via sonication for 15 min, and equilibrated to 37 ± 0.5°C prior to testing.
Method validation
Metformin standard stock solutions (100 µg/mL) were serially diluted to prepare calibration curves for each dissolution medium in the range of 2–20 µg/mL. The calibration curves displayed excellent linearity with R2 values consistently >0.999. Accuracy was assessed via recovery studies at 80%, 100%, and 120% concentration levels, yielding recoveries between 98.5% and 101.2%. Intra-day and inter-day precision studies showed %RSD <2%, indicating method reliability. Limit of Detection (LOD) and Limit of Quantification (LOQ) were calculated using the standard deviation of the response and slope method, confirming the method’s sensitivity.26–37
Dissolution testing procedure
Dissolution testing was conducted using the USP Apparatus II (paddle method) in accordance with FDA and WHO biowaiver guidelines as follows: Number of tablets per brand: Six units (n = 6) per medium Medium Volume: 900 mL per vessel Paddle speed: 50 revolutions per minute (rpm) Temperature: Maintained at 37 ± 0.5°C throughout the test Time Intervals: 5, 10, 15, 30, 45, and 60 min
At each interval, 10 mL of the dissolution medium was withdrawn using a syringe fitted with a 0.45 µm membrane filter. An equal volume of fresh, pre-warmed dissolution medium was added to maintain constant volume and sink conditions. The filtered samples were analyzed immediately using a UV-Vis spectrophotometer at 233 nm (validated wavelength for metformin detection). A blank dissolution medium was used for baseline correction.38–42
Data analysis
The cumulative percentage of drug release was calculated for each time point using Beer-Lambert’s law. Data from triplicate runs were averaged and presented with standard deviation to assess similarity and bioequivalence:
15
• Difference Factor (f1): • Similarity Factor (f2): • Dissolution Efficiency (DE):
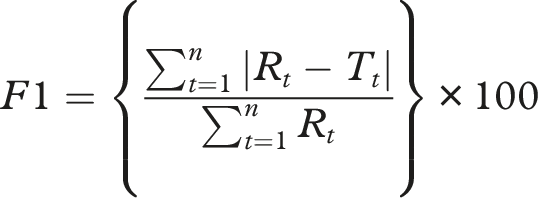
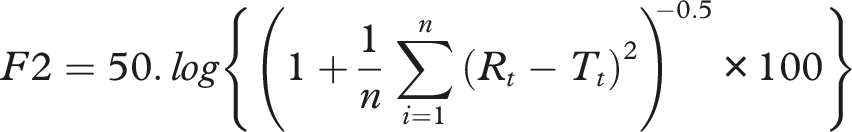
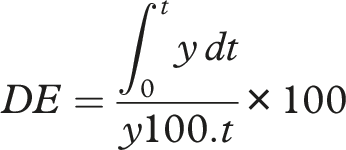
Results and discussion
Dissolution profiles across pH conditions
The findings of this comparative in-vitro dissolution study highlight important differences and similarities among commercially available metformin hydrochloride 500 mg tablet formulations. This evaluation is crucial, given metformin’s status as a Biopharmaceutics Classification System (BCS) Class III drug, where bioequivalence can potentially be waived if in-vitro dissolution similarity is established across a range of physiological pH levels.43,44
BCS Class III drugs, including metformin hydrochloride, are characterized by high solubility but low permeability. The primary concern with such drugs lies not in dissolution but in their potential site-dependent absorption in the gastrointestinal tract. Therefore, regulatory bodies such as the FDA, EMA, and WHO emphasize the necessity for rapid and similar dissolution profiles in three pH conditions: 1.2 (gastric), 4.5 (early intestinal), and 6.8 (late intestinal).45,46
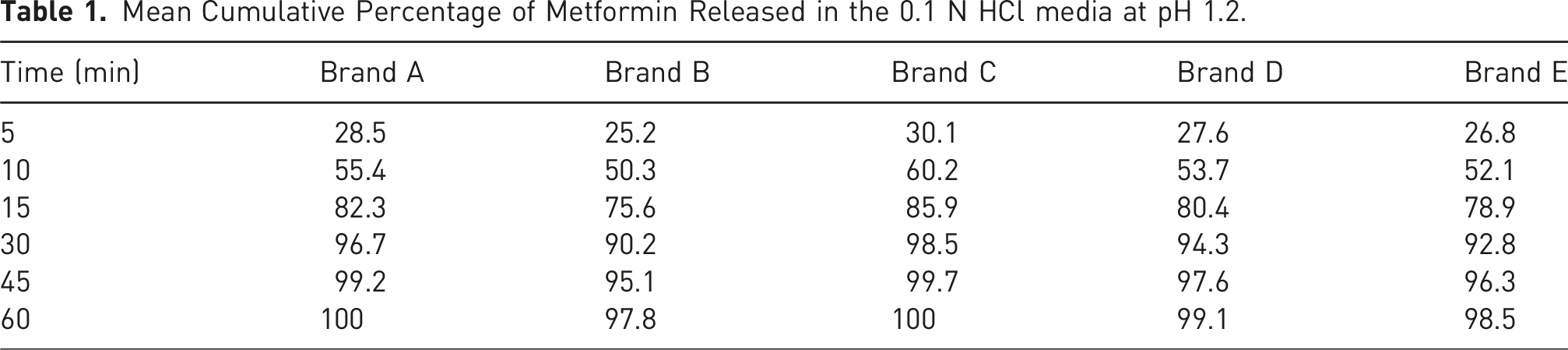
Mean Cumulative Percentage of Metformin Released in the 0.1 N HCl media at pH 1.2.
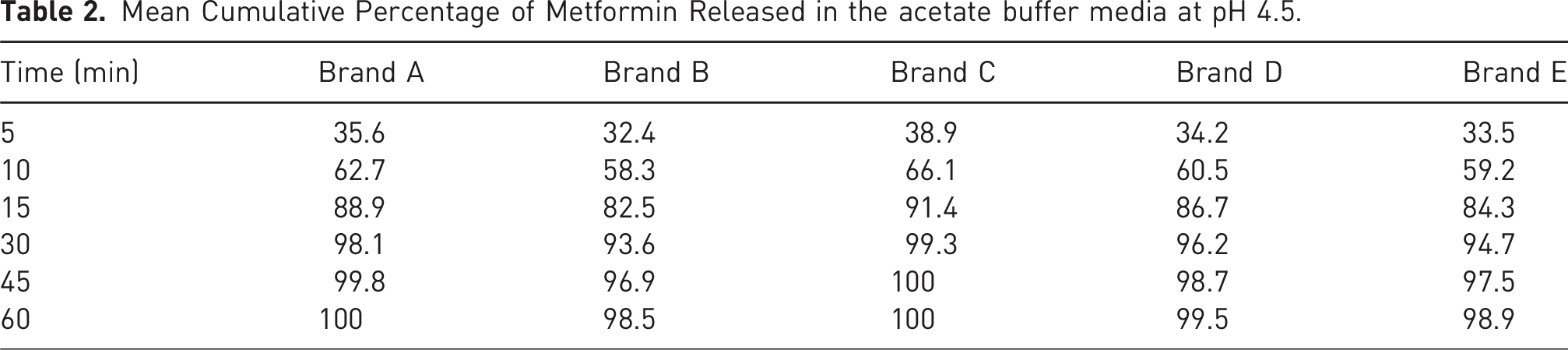
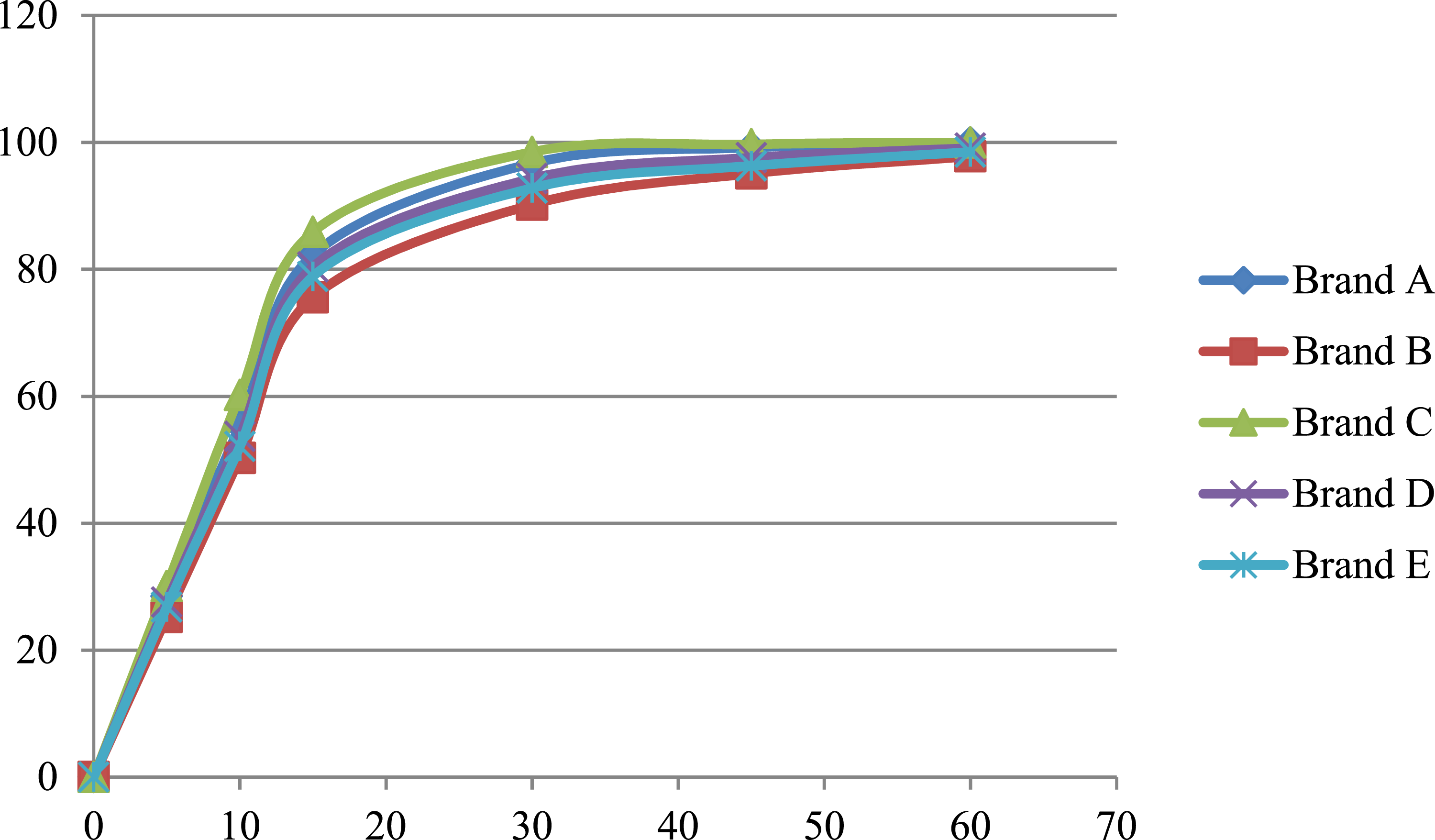
Mean Cumulative Percentage of Metformin Released in the acetate buffer media at pH 4.5.
Mean Cumulative Percentage of Metformin Released in the Phosphate buffer media at pH 6.8.
Mean Cumulative Percentage of Metformin Released in the 0.1 N HCl media at pH 1.2.
Mean Cumulative Percentage of Metformin Released in the acetate buffer media at pH 4.5.
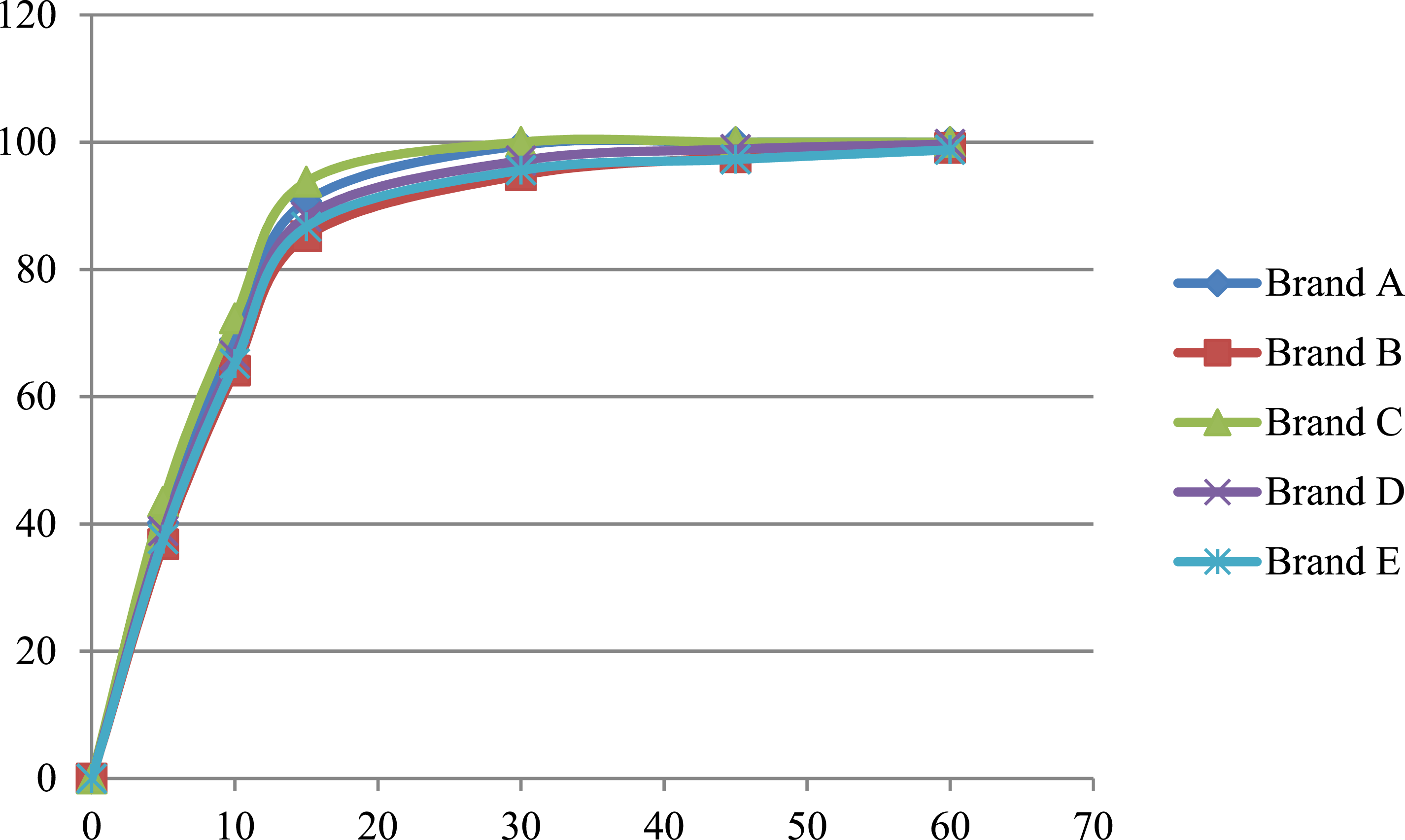
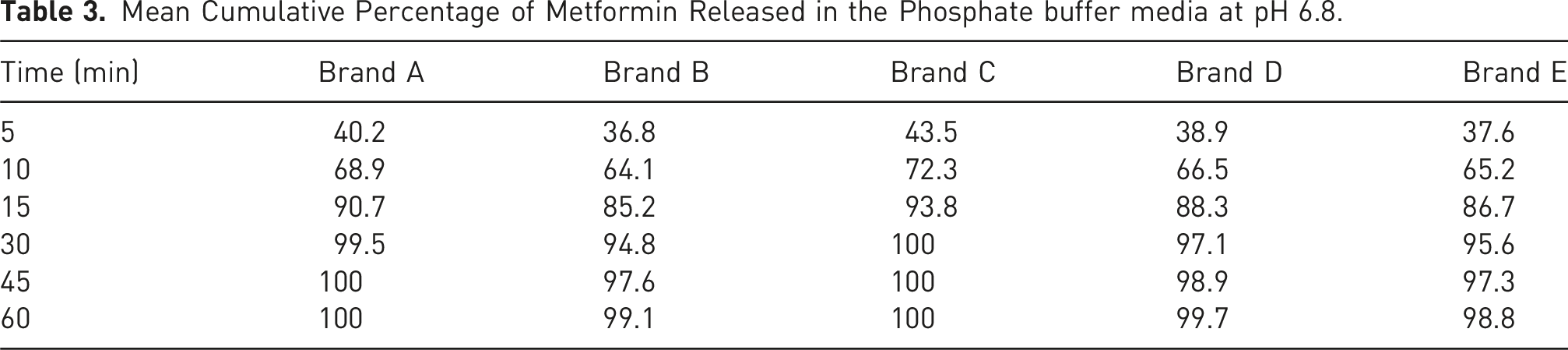
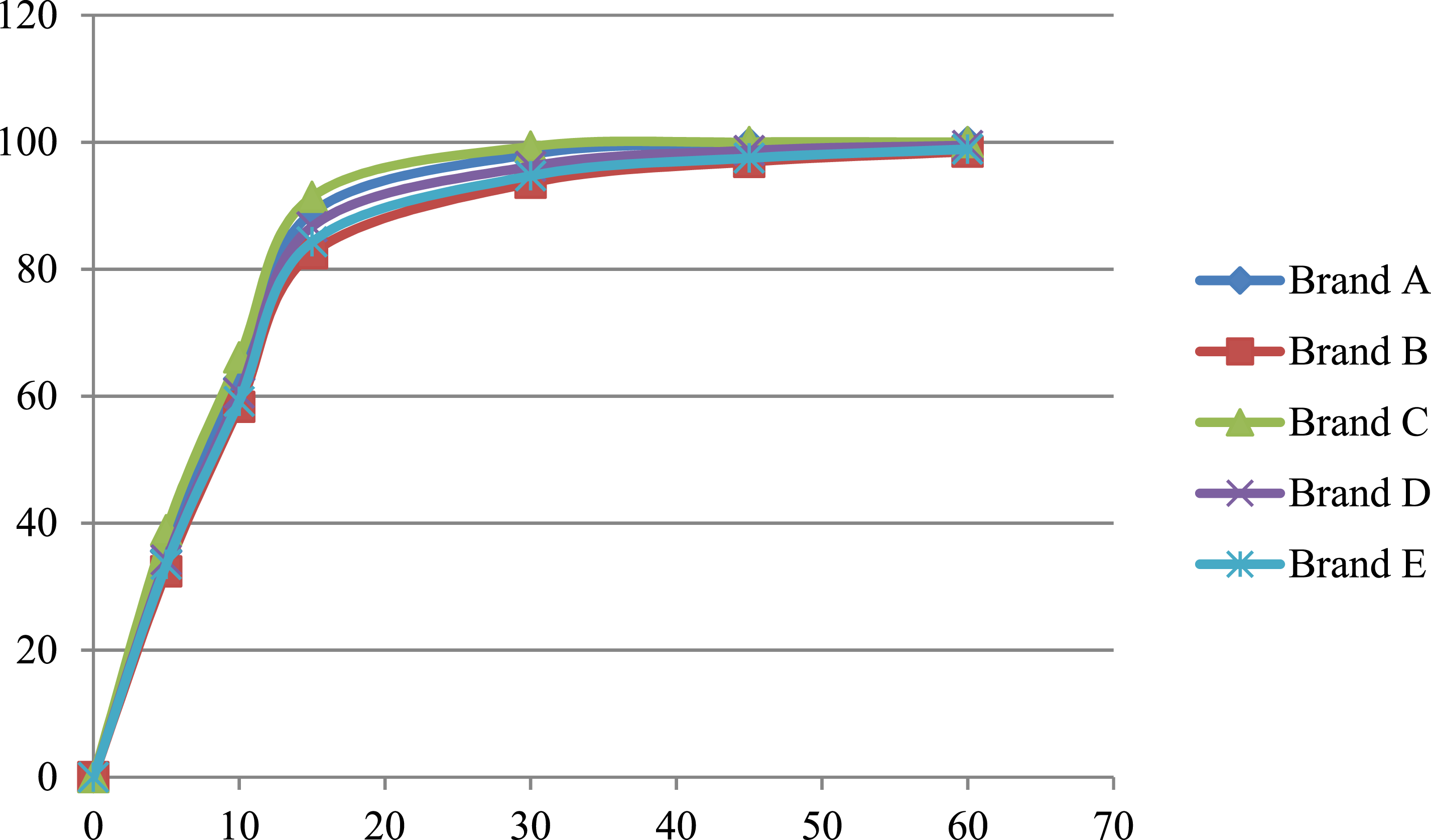
Mean Cumulative Percentage of Metformin Released in the Phosphate buffer media at pH 6.8.
Comparative analysis using f1 and f2 factors
f1 and f2 Values Comparing Test Brands with to Reference to Brand C.
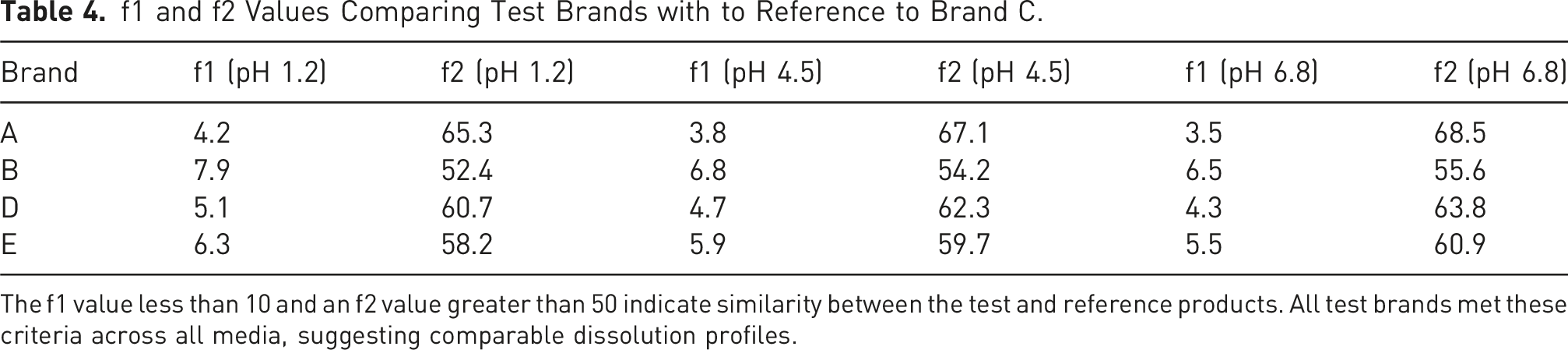
The f1 value less than 10 and an f2 value greater than 50 indicate similarity between the test and reference products. All test brands met these criteria across all media, suggesting comparable dissolution profiles.
Dissolution efficiency (DE)
Dissolution efficiency (%) at 60 Min.
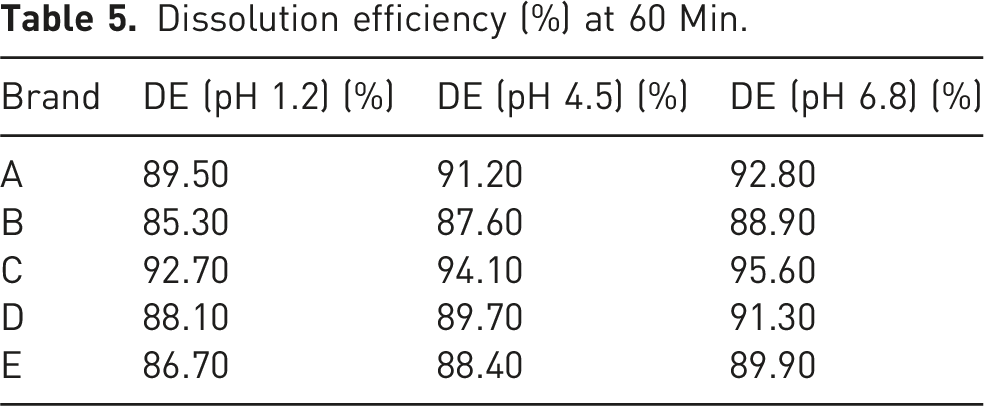
Brand C exhibited the highest dissolution efficiency across all media, aligning with its role as the reference product. Brand C was used as the reference due to its consistent performance, Brands A and D also showed acceptable similarity (f2 > 50) and high dissolution efficiency values. These findings suggest that these brands can be considered pharmaceutically equivalent to the reference. However, Brands B and E, though falling within acceptable ranges, demonstrated slightly lower dissolution efficiency values and higher f1 values, indicating possible batch-to-batch variability or formulation issues.
These results repeat findings from several recent studies, such as Bhaskarabhatla A et al. (2017), who observed considerable inter-brand differences in dissolution behavior among metformin tablets sourced from different manufacturers across the country. 47 Similarly, Bhushan R et al. (2024) reported that while Indian generics met USP dissolution standards, few achieved bioequivalence when f2 values were rigorously applied. 48
The observed variability among formulations is likely attributable to differences in excipients and manufacturing practices. For BCS Class III drugs, excipients that influence gastrointestinal transit or interact with the drug to form complexes may affect absorption and ultimately bioavailability, even if the drug is highly soluble. 49 Hydrophilic excipients such as sodium starch glycolate or crospovidone tend to enhance disintegration and dissolution rates, whereas hydrophobic fillers or binders may retard drug release. 50
These factors were likely at play in the underperformance of Brand E, which, despite releasing more than 85% of drug within 15 min, showed irregular dissolution curves and lower f2 values. This inconsistency underscores the importance of stringent excipient controls during formulation development, especially for biowaiver submissions.
The study’s findings have significant implications for regulatory approvals, clinical practice, and patient outcomes. Regulatory authorities rely heavily on in-vitro data for approving BCS Class III generic formulations without requiring human bioequivalence studies. Inconsistencies like those seen in Brands B and E could result in therapeutic failures or unexpected pharmacokinetic variability if not adequately controlled.
Clinical equivalence is especially critical for metformin, which is often prescribed chronically and used in combination therapies. Fluctuations in its release or absorption could affect glycemic control, increasing the risk of complications in diabetic patients. 51 It is therefore essential that manufacturers ensure formulation robustness and that national regulatory bodies conduct regular post-marketing surveillance.
A 2023 meta-analysis emphasized that even among approved generics, 12% failed to maintain consistent dissolution profiles under quality reassessment conditions, further highlighting the need for vigilance in this space. 52
In Low- and Middle-Income Countries (LMICs), generic medications represent the bulk of pharmaceutical consumption. Ensuring that these generics conform to international biowaiver standards is crucial for public health. The WHO has published guidance recommending rigorous in-vitro dissolution testing as a prerequisite for granting waivers, especially where the infrastructure for in-vivo testing is limited. 53
Studies in Nigeria and Bangladesh have shown that up to 30% of marketed generics of essential medicines like metformin do not meet WHO biowaiver criteria, often due to poor regulatory oversight and weak pharmaceutical quality assurance systems.54,55
While the FDA, EMA, and WHO provide similar criteria for granting biowaivers, minor variations still exist regarding buffer volumes, test conditions, and statistical thresholds. Harmonizing these standards could improve regulatory efficiency and product interchangeability across markets. In the context of the growing global burden of type 2 diabetes, such harmonization is no longer optional but essential. 56
Conclusion
This study undertook a comprehensive comparative in-vitro dissolution evaluation of five commercially available brands of metformin hydrochloride 500 mg tablets using established biowaiver protocols under simulated gastrointestinal conditions. The central aim was to assess the interchangeability of these generic formulations by analyzing their dissolution profiles in three different pH media (pH 1.2, 4.5, and 6.8) and applying regulatory metrics such as similarity factor (f2), difference factor (f1), and dissolution efficiency (DE). These parameters serve as critical tools for determining whether a generic formulation qualifies for a biowaiver-based approval pathway, eliminating the need for costly and time-consuming in-vivo bioequivalence studies. Brand C exhibited the highest dissolution efficiency across all media, aligning with its role as the reference product. In this study, almost all five formulations released more than 85% of the drug within 15 min under the three tested conditions, suggesting compliance with the “very rapidly dissolving” criterion set forth by regulatory authorities. However, variations in f1, f2, and dissolution efficiency metrics among brands indicate differences in formulation technology, excipient selection, and manufacturing consistency. From a regulatory perspective, this study reaffirms the viability and value of in-vitro dissolution testing as a surrogate for in-vivo bioequivalence, particularly for BCS Class III compounds. Regulatory bodies such as the FDA, EMA, and WHO provide clear guidelines that, when properly followed, enable generic manufacturers to gain approval more efficiently while maintaining patient safety and therapeutic efficacy. However, our findings also caution against blanket substitution of generics without confirmatory dissolution data, as not all marketed formulations demonstrate equivalence to the innovator product. The use of non-equivalent generics may also erode patient confidence in generic substitution, which is vital for healthcare systems reliant on cost-effective medication. Future work should extend beyond in-vitro testing to include in-vivo–in-vitro correlation (IVIVC) studies, real-world therapeutic outcomes, and broader market assessments to ensure safe, effective, and affordable access to essential medications like metformin.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.