
Abstract
Aβ-afferents are traditionally believed to be low-threshold mechanoreceptors (LTMRs) involved in sensing innocuous mechanical stimuli such as gentle touch; however, emerging evidence indicates that a subset of Aβ-afferents functions as high-threshold mechanoreceptors (HTMRs) involved in mechanical pain and termed Aβ-afferent nociceptors. While cooling is known to usually suppress neuronal excitability, its effects on the excitability of Aβ-afferent nociceptors remain unclear. Here, we used Nav1.8ChR2/eYFP mice and patch-clamp recordings to examine how cooling affects intrinsic membrane properties, excitability, and ionic currents in Aβ-afferent nociceptors in the trigeminal ganglia (TG). Aβ-afferent nociceptors were identified as Nav1.8-positive (eYFP-positive) large-diameter (34–43 μm) TG neurons with their axon conduction velocities >13 m/s at 33°C. Cooling from 33 to 10°C progressively reduced axon conduction velocity and increased their stimulation threshold. However, at the soma of Aβ-afferent nociceptors, cooling decreased the action potential (AP) rheobase, increased AP amplitude and input resistance, and depolarized the resting membrane potential, although it increased the AP threshold and width. Voltage-clamp recordings showed that cooling suppressed both inward Na+ and outward K+ currents at the soma. Interestingly, the cooling-induced reduction in outward K+ currents was largely mediated by temperature-sensitive two-pore-domain potassium (K2P) channels at the soma, suggesting a role in shifting intrinsic membrane properties toward increased excitability. Enhancing intrinsic membrane excitability at the soma of Aβ-afferent nociceptors raises the possibility that cooling may similarly affect excitability at their endings.
Keywords
Introduction
Emerging evidence reveals that a subpopulation of Aβ-afferents functions as high-threshold mechanoreceptors (HTMRs) involved in mechanical pain. 1 Classically, Aβ-afferents are believed to be primarily low-threshold mechanoreceptors (Aβ-afferent LTMRs) that convey innocuous mechanical stimuli, such as gentle touch. 2 In contrast, Aβ-afferent HTMRs only respond to noxious mechanical stimuli and thereby are regarded as Aβ-afferent nociceptors. 1 Aβ-afferent nociceptors have been found to comprise 18%–65% of cutaneous nociceptors across different species, including mice, rats, guinea pigs, cats, and monkeys. 1 Aβ-afferent nociceptors are distinguished from Aδ- and C-afferent nociceptors by their faster conduction velocities, serving as first responders to harmful mechanical stimuli.1,3 Aβ-afferent nociceptors have been identified in rat trigeminal afferents, showing chemical sensitivity but displaying minimal mechanically activated (MA) currents. 4 Under pathological conditions, such as inflammation and nerve injury, Aβ-afferent nociceptors are sensitized to contribute to the development of mechanical allodynia.3,5,6
Aβ-afferent nociceptors exhibit molecular features typically associated with classical afferent nociceptors. For example, Aβ-afferent nociceptors express Nav1.8,5,6 a tetrodotoxin-resistant voltage-gated Na+ channel that is primarily expressed in C-afferent nociceptors.7,8 Nav1.8 plays a key role in the initiation and propagation of APs in classical afferent nociceptors. 7 Its depolarized activation and slow inactivation enable a sustained inward current that supports AP generation even when TTX-sensitive voltage-gated Na+ channel subtypes are pharmacologically blocked. 7 Importantly, compared to TTX-sensitive voltage-gated Na+ channels, the activity of Nav1.8 channels is less suppressed at low temperatures, thereby supporting AP generation in classical afferent nociceptors at noxious cooling temperatures. 9 Nav1.8 activity may also be retained at low temperatures in Aβ-afferent nociceptors, thereby supporting AP firing in Aβ-afferent nociceptors. At low temperatures, the ability of Aβ-afferent nociceptors to fire APs can be important for rapidly detecting and avoiding mechanical injury.
Cooling can affect somatosensory functions through multiple mechanisms. Cooling can directly excite subpopulations of C-afferents by activating TRPM8 channels, thereby inducing either an innocuous cooling sensation or noxious cold pain.10–12 Cooling can also modulate afferent membrane excitability, independent of TRPM8 channels, thereby influencing somatosensory perception. For example, cooling can suppress Aβ-afferent LTMR excitability and reduce their mechanical sensitivity. 13 Mechanistically, in afferent neurons without TRPM8 channels, the effects of cooling on membrane excitability may be due to the suppression of ion channel activity, which in turn alters the intrinsic membrane properties of afferents such as Aβ-afferent LTMRs. In addition to Aβ-afferent LTMRs, cooling temperatures have been shown previously to differentially affect intrinsic membrane properties and AP firing in C-afferent TG neurons. 14 However, the effects of cooling on the intrinsic membrane properties, excitability, and ion channel activity in Aβ-afferent nociceptors remain not determined.
Two-pore domain potassium (K2P) channels, which mediate background leak K+ currents, are critically involved in controlling intrinsic membrane properties in neuronal cells. 15 Of 15 K2P channels identified in mammals, TREK1, TREK2, and TRAAK are highly temperature-sensitive, and their activity can be significantly reduced at cooling temperatures.16,17 However, a controversial finding has been reported for TRAAK. 18 Cooling temperatures may suppress the activity of temperature-sensitive K2P channels in afferent neurons, thereby altering their intrinsic membrane properties and excitability if these channels are expressed there. However, it is currently unknown whether temperature-sensitive K2P channels are expressed in Aβ-afferent nociceptors and, if so, how the activity of K2P channels is affected to contribute to the changes in the intrinsic membrane properties and excitability of Aβ-afferent nociceptors at cooling temperatures. In addition to the temperature-sensitive K2P channels, cooling may suppress the activity of voltage-gated Na+ and K+ channels, thereby contributing to changes in the intrinsic membrane properties and excitability in Aβ-afferent nociceptors.
In the present study, we examined the effects of cooling temperatures on conduction and intrinsic membrane properties in trigeminal Aβ-afferent nociceptors using patch-clamp recordings. We also assessed the effects of cooling temperatures on inward currents mediated by voltage-activated Na+ channels and outward currents mediated by temperature-sensitive K2P channels. We further used immunostaining and confocal microscopy to determine the expression of temperature-sensitive K2P channels in trigeminal Aβ-afferent nociceptors.
Materials and methods
Animals
Nav1.8ChR2/eYFP mice were used in this study. Nav1.8ChR2/eYFP mice were generated by crossing Scn10a-Cre (Nav1.8-Cre) and Ai32 (RCL-ChR2(H134R)/eYFP) transgenic mice. Scn10a-Cre mice were gifts from Dr. John Wood at University College London and transferred to us from Dr. Stephen Waxman’s lab at Yale University. Ai32 mice were purchased from Jackson Labs. We performed a crossbreeding of Nav1.8-Cre mice with Ai32 (RCL-ChR2(H134R)/eYFP) mice to establish the Nav1.8-Cre+; ChR2-eYFP loxP/+ mouse line, referred to as Nav1.8ChR2/eYFP hereafter. These mice express eYFP in their Nav1.8-positive (Nav1.8+) TG neurons, which are mainly nociceptors. The mice used in the present study were aged 13–21 weeks, and both males and females were included. All animals were housed in a temperature-controlled room at 23°C and maintained on a 12 h light/dark cycle. All animal care and experimental procedures were conducted following the guidelines established by the National Institutes of Health (NIH) for the care and use of experimental animals. Approval of the experimental protocols employed in this study was granted by the Institutional Animal Care and Use Committee (IACUC) at the University of Alabama at Birmingham.
Ex vivo trigeminal ganglion preparations and patch-clamp recordings
Ex vivo TG preparations and patch-clamp recordings followed methods from our earlier work.4,14 In brief, Nav1.8ChR2/eYFP mice were anesthetized and decapitated, and trigeminal ganglia (TG) were bilaterally dissected out with attached infraorbital (V2) nerve bundles (5–7 mm) and submerged in ice cold Krebs solution in a 35 mm petri dish containing (in mM): 117 NaCl, 3.6 KCl, 1.2 Na2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 25 glucose. The Krebs solution was saturated with 95% O2 and 5% CO2, had a pH of 7.35, and an osmolarity of 324 mOsm. After removal of superficial connective tissue, the ex vivo TG was placed in a recording chamber with the ventral surface facing upward, secured with a tissue anchor, and perfused with Krebs solution at 23°C. The recording chamber was mounted on the stage of an Olympus BX51 microscope equipped with a fluorescence imaging system. TGs were treated with 0.06% dispase II (Roche, Indianapolis, IN, USA) and 0.06% collagenase (MilliporeSigma, Billerica, MA, USA) in Krebs solution for ~6 min at 23°C, followed by washout and continuous perfusion with Krebs solution at 2 ml/min. For patch-clamp recordings, electrodes were fabricated using a micropipette puller (Sutter Instruments). The electrode resistance ranged from 4 to 6 MΩ after filling the recording electrode internal solution. The recording electrode internal solution contained (in mM): 105 K-gluconate, 35 KCl, 0.5 CaCl2, 2.4 MgCl2, 5 EGTA, 10 HEPES, 5 Na2ATP, and 0.33 GTP-TRIS salt; the pH was adjusted to 7.35 with KOH. The junction potential was 12 mV, calculated from the ionic concentrations of the internal and bath solutions using pCLAMP 11 (Molecular Devices). Recordings were conducted on Nav1.8-positive (Nav1.8+, eYFP-positive). Nav1.8+ Aβ-afferent TG neurons were identified using fluorescence microscopy with an eYFP/GFP filter set (Excitation: 470–490 nm; Emission: 520–550 nm), in combination with conduction velocity (CV) measurement. After establishing whole-cell access, recordings were performed in current-clamp mode first to classify TG neurons based on the AP conduction velocity (CV) of their afferent fibers. APs were evoked by applying electrical stimulation at the peripheral end of afferent nerve bundles using a suction stimulation electrode. The suction stimulation electrode was fire-polished and had a tip diameter of ∼1 mm. The peripheral end of the afferent nerve fibers was aspirated into the suction stimulation electrode under negative pressure. APs were evoked by monophasic square wave pulses generated by pClamp11 software and delivered via a stimulation isolator (ISO-Flex, A.M.P.I.). The stimulation pulse duration was 50 µs. CV was calculated from AP latency and afferent nerve length. The latency of APs was measured from the time of stimulation, which was marked by a stimulation artifact, to the time when AP was initiated at the recorded TG neuron. The length of the afferent nerves between the site of electrical stimulation and the recording site was 5–7 mm. The afferent fibers with CV ≥10 m/s at 23°C were considered Aβ-afferents (25). To determine the properties of membranes and APs of recorded TG neurons, patch-clamp recordings were performed on the soma under the whole-cell current-clamp configuration. Step currents were injected into TG neurons through recording electrodes. Step currents were applied from −100 pA to 3000 pA in 100 pA increments, with a 500-ms step duration. APs evoked by current steps were used to determine AP properties, including AP rheobase, amplitude, width, and threshold. To characterize voltage-activated ionic currents in TG neurons, whole-cell patch-clamp recordings were performed in voltage-clamp mode, with the membrane held at −72 mV. Voltage steps ranged from −102 mV to 38 mV (command voltages from −90 mV to 50 mV) in 10 mV increments, with each step lasting 500 ms. Unless otherwise stated, all reported membrane voltages were corrected for a calculated junction potential of 12 mV. Signals were amplified with an Axopatch 200B amplifier, low-pass filtered at 5 kHz, and digitized at 10 kHz. All recordings were acquired using pCLAMP 11 software from Molecular Devices. Unless otherwise stated, recordings were conducted at temperatures of 33°C, 23°C, 15°C, and 10°C, which were achieved by using a Warner Instruments CL-200 heater-cooler controller.
Pharmacology
BaCl₂ (5 mM) and norfluoxetine (250 μM) were used to inhibit K2P channels (TREK1, TREK2, and TRAAK) and were prepared in Krebs solution. Both agents were bath-applied to TG neurons for 10 min prior to recording. Voltage-gated K+ (Kv) channel blockers included CsCl (135 mM), 4-aminopyridine (4-AP; 1 mM), and tetraethylammonium (TEA; 10 mM). CsCl was included in the internal solution, replacing K-gluconate and KCl, to achieve intracellular blockade. 4-AP and TEA were bath-applied for 10 min prior to recording.
Data analysis
Electrophysiological recordings were obtained from Nav1.8+ Aβ-afferent TG neurons collected across multiple experimental sessions from approximately 30 male and 30 female animals. Since we did not observe any obvious differences in electrophysiological data between males and females, data from both sexes were combined for analysis. To minimize sampling bias, recordings were obtained from multiple independent preparations on different days, and 2–3 neurons were usually recorded from each TG. Individual neurons were treated as independent experimental units for electrophysiological data analyses. Only recordings with access resistance < 25 MΩ were included for data analysis. Recordings were excluded if access resistance changed by more than 20%, or baseline currents exceeded −100 pA at the holding voltage of −72 mV during experiments. For voltage-clamp recording data, the area under the curve (AUC) was calculated from current-voltage (I-V) curves of voltage-activated currents. Data are presented as mean ± SEM unless otherwise stated. Statistical analyses were performed using one-way ANOVA with post hoc Šídák’s multiple comparisons test, with statistical significance set at p < 0.05.
Results
Ex vivo TG preparations were made from Nav1.8ChR2/eYFP mice, and patch-clamp recordings were performed selectively in large-sized eYFP-positive, that is, Nav1.8+, TG neurons (Figure 1(a)–(c)) as we aimed to study trigeminal Aβ-afferent nociceptors. The diameters of these large-sized Nav1.8+ TG neurons ranged from 34 to 43 μm (36.5 ± 0.3 μm, n = 52, Figure 1(c)). We first examined the AP conduction velocity (CV) along the axons of Nav1.8+ Aβ-afferents at temperatures of 33°C–10°C. Cooling progressively slowed CV in Nav1.8+ Aβ-afferent axons (Figure 1(d) and (e)). CV was 15.6 ± 0.4 m/s (n = 19) at 33°C, significantly decreased to 11.6 ± 0.3 m/s (n = 27) at 23°C, 7.8 ± 0.4 m/s (n = 17) at 15°C, and 5.3 ± 0.5 m/s (n = 15) at 10°C (Figure 1(e)). Electrical stimulation thresholds required to evoke APs from the afferent nerve bundles were 347.1 ± 30.8 μA (n = 14) at 33°C, were not significantly affected at 23°C (453.6 ± 33.7 μA, n = 14), significantly increased to 1054 ± 62.6 μA (n = 13) at 15°C, and 3611 ± 462.3 μA (n = 9) at 10°C (Figure 1(f)). At the soma of Nav1.8+ Aβ-afferent TG neurons, membrane capacitance did not differ significantly across temperatures tested (Figure 1(g)). Input resistance was 23.7 ± 1.4 MΩ (n = 25) at 33°C, 34.2 ± 2.0 MΩ (n = 39, p = 0.09, no significant difference) at 23°C, significantly increased to 106.1 ± 7.5 MΩ (n = 14) at 15°C, and 132.7 ± 8.4 MΩ (n = 14) at 10°C (Figure 1(h)). Resting membrane potentials (RMP) were −85.0 ± 0.6 mV (n = 28) at 33°C, −83.1 ± 0.7 mV (n = 46, no significant difference) at 23°C, significantly less negative at 15°C (−76.8 ± 2.4 mV, n = 14) and at 10°C (−75.4 ± 1.3 mV, n = 14) compared with at 33°C (Figure 1(i)).
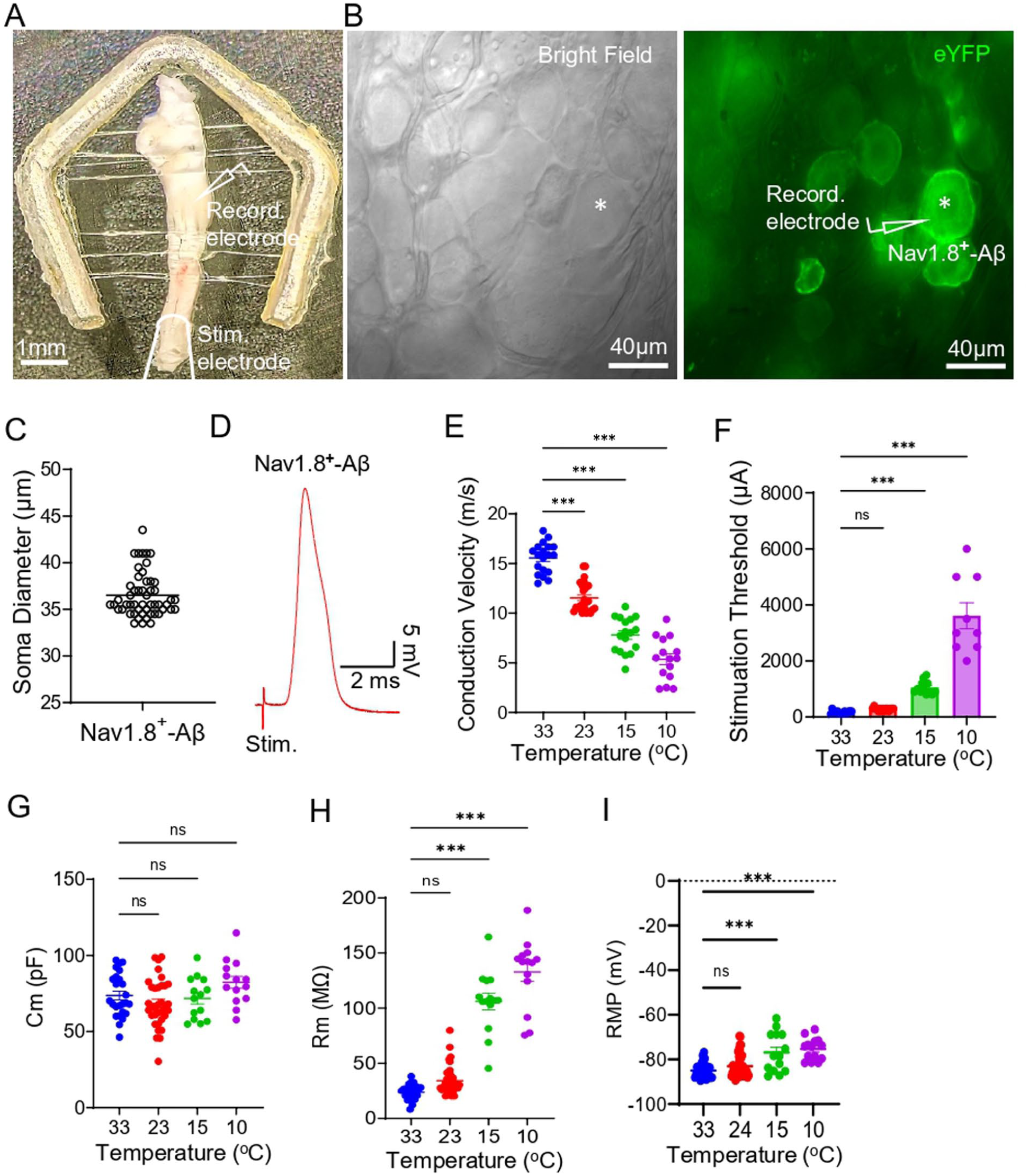
Effects of cooling on action potential conduction velocity and passive membrane properties of Nav1.8+ Aβ-afferent TG neurons. (a) Ex vivo TG preparations and the illustration of the site (V2 region) of patch-clamp recordings and the site of electrical stimulation. (b) Bright-field (left panel) and eYFP fluorescence (right panel) images show individual TG neurons, including a Nav1.8+ Aβ-afferent neuron (asterisk indicated), viewed under a 40x objective. (c) Soma diameters of the recorded Nav1.8+ Aβ-afferent TG neuron. (d) Sample Trace shows an AP recorded from a Nav1.8+ Aβ-afferent TG neuron following electrical stimulation of the afferent nerve bundle. (e) Afferent conduction velocities measured at 33°C, 23°C, 15°C, and 10°C for the recorded Nav1.8+ Aβ-afferent TG neurons. (f) Stimulation threshold to evoke action potentials in the afferent nerve bundles at 33°C, 23°C, 15°C, and 10°C. (g–i) Membrane capacitance (Cm, G), input resistance (Rm, H), and resting membrane potentials (RMP, I) of the recorded Nav1.8+ Aβ-afferent TG neurons at 33°C, 23°C, 15°C, and 10°C. Data are presented as mean ± SEM. One-way ANOVA with post hoc Šídák’s multiple comparisons test, ns, not significant, *** p < 0.001.
We investigated how cooling temperatures affect active membrane properties on the soma of Nav1.8+ Aβ-afferent TG neurons (Figure 2). APs were elicited from Nav1.8+ Aβ-afferent TG neurons by direct injection of depolarizing currents to these cells at 33°C, 23°C, 15°C, and 10°C (Figure 2(a)). At 33°C, AP rheobase, the threshold current required to elicit an AP, was high at 2768 ± 143 pA (n = 22), significantly decreased to 1597 ± 55 pA (n = 35) at 23°C, 846 ± 46 pA, (n = 13) at 15°C, and 700 ± 52 pA (n = 13) at 10°C (Figure 2(b)). AP threshold was −32 ± 2 mV (n = 22) at 33°C and was not significantly affected at cooling to 23°C and 15°C but became significantly less negative at 10°C (−16 ± 3 mV, n = 13; Figure 2(c)). AP width was 0.9 ± 0.1 ms (n = 22) at 33°C, 1.4 ± 0.1 ms (n = 36, no significant difference) at 23°C but significantly broadened to 3.6 ± 0.3 ms (n = 14) at 15°C and 7.1 ± 0.8 ms (n = 14) at 10°C. AP amplitude was 113 ± 3 mV (n = 20) at 33°C, not significantly affected at 23°C (n = 33), but was significantly increased to 122 ± 1 mV (n = 13) at 15°C and 125 ± 1 mV (n = 13) at 10°C.
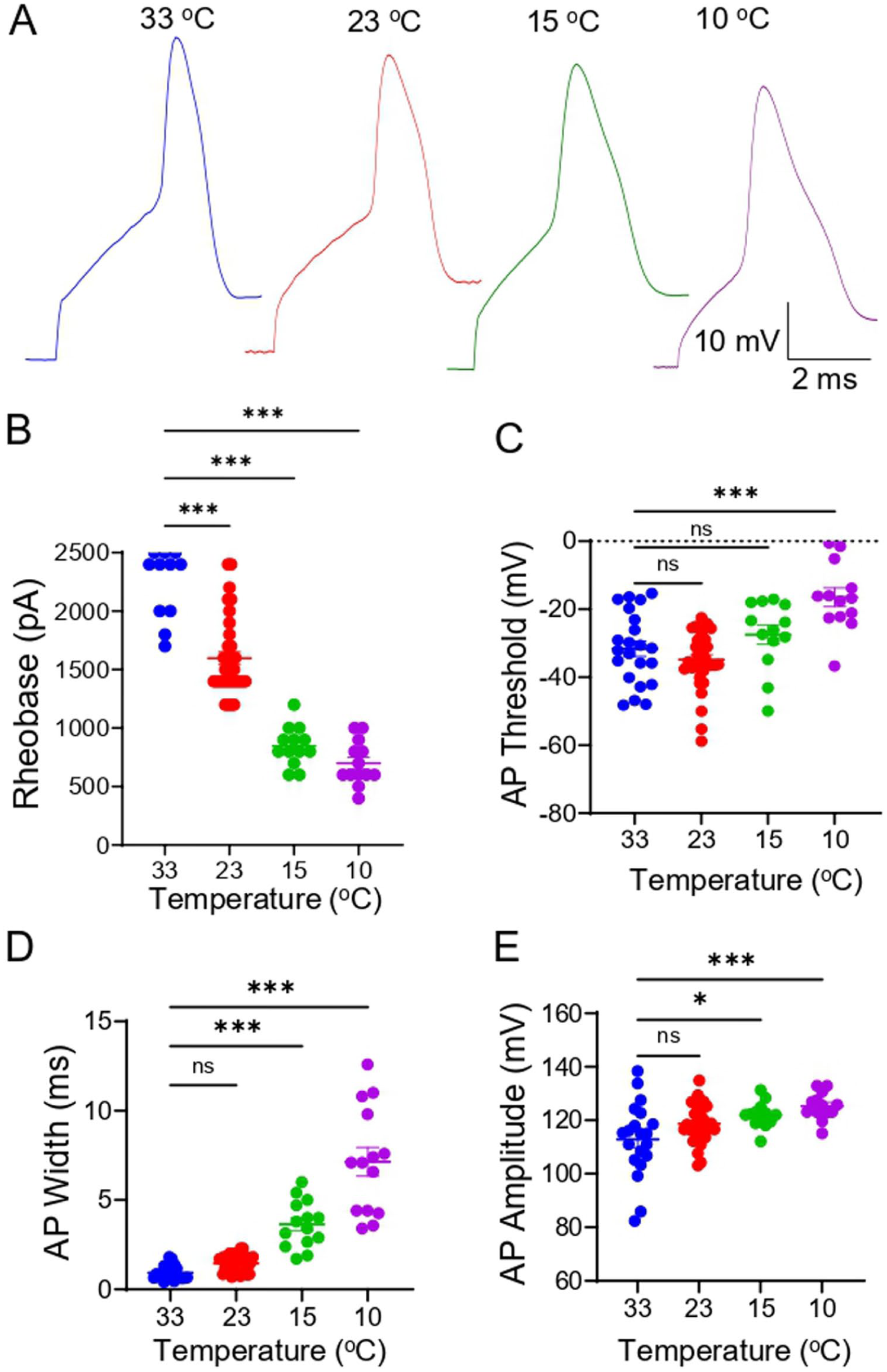
Effects of cooling on active membrane properties of Nav1.8+ Aβ-afferent TG neurons. (a) Sample traces of APs elicited from Nav1.8+ Aβ-afferent TG neurons by direct injection of depolarizing currents at 33°C (1st), 23°C (2nd), 15°C (3rd), and 10°C (4th). (b–e) AP Rheobase (b), AP threshold (c), AP width (d), and AP amplitude measured at 33°C, 23°C, 15°C, and 10°C. Data are presented as mean ± SEM. One-way ANOVA with post hoc Šídák’s multiple comparisons test, ns, not significant, *p < 0.05, ***p < 0.001.
We investigated the effects of cooling on voltage-activated currents in the soma of Nav1.8+ Aβ-afferent TG neurons. In voltage-clamp mode, voltage steps evoked transient inward currents mediated by voltage-gated Na+ channels and sustained outward currents mediated by K+ channels in Nav1.8+ Aβ-afferent TG neurons (Figure 3(a)). Cooling from 33°C to 10°C progressively reduced inward currents, as evidenced by decreased current amplitudes across depolarizing voltage steps (Figure 3(a) and (b)). Quantitative analysis of the area under the I-V curve (AUC) of inward currents showed a significant reduction with decreasing temperature, from 422 ± 34 nA × mV (n = 17) at 33°C to 335 ± 15 nA × mV (n = 29) at 23°C, 236 ± 17 nA × mV (n = 15) at 15°C, and 108 ± 9 nA × mV (n = 15) at 10°C (Figure 3(c)). Thus, over 25% of inward currents remained even at 10°C (Figure 3(b) and (c)). Outward K+ currents also exhibited temperature dependence (Figure 3(d)). The AUC of initial outward current component was 409 ± 20 nA × mV (n = 19) at 33°C, not significantly affected (415 ± 24 nA × mV, n = 26) at 23°C, but significantly reduced to 282 ± 22 nA × mV (n = 15) at 15°C, and 172 ± 11 nA × mV (n = 15) 10°C (Figure 3(e)). Similarly, the late outward current component showed significant suppression at 15°C (n = 15) and 10°C (n = 14; Figure 3(g)).
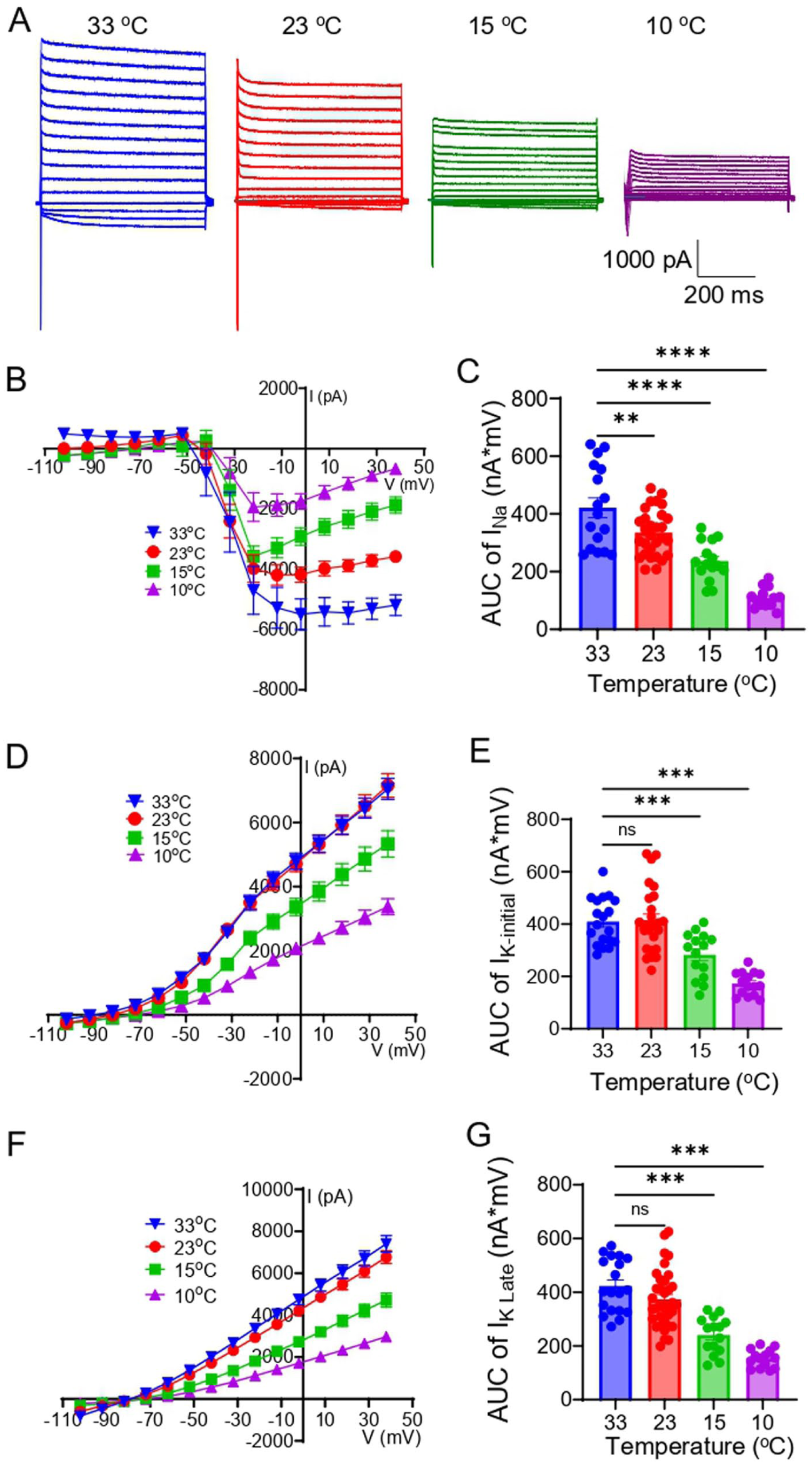
Effects of cooling on voltage-activated currents in Nav1.8+ Aβ-afferent TG neurons. (a) Sample traces of voltage-activated inward and outward currents measured at 33°C, 23°C, 15°C, and 10°C in Nav1.8+ Aβ-afferent TG neurons. (b) I-V Curve of voltage-activated inward currents at 33°C, 23°C, 15°C, and 10°C. (c) AUC of voltage-activated inward currents at 33°C, 23°C, 15°C, and 10°C. (d) I-V curve of voltage-activated outward currents in the initial phase at 33°C, 23°C, 15°C, and 10°C. (e) AUC of the voltage-activated outward currents in the initial phase at 33°C, 23°C, 15°C, and 10°C. (f–g) Similar to D&E, except that the voltage-activated outward currents in the late phase were shown. Data are presented as mean ± SEM. One-way ANOVA with post hoc Šídák's multiple comparisons test, ns, not significant, **p < 0.01, ***p < 0.001.
Suppressive effects of cooling temperatures on total outward currents in Nav1.8+ Aβ-afferent TG neurons (Figure 3(d)–(g)) might be partly due to the inhibition of temperature-sensitive leak K+ channels. Therefore, we examined whether temperature-sensitive leak K+ channels, including TREK1, TREK2, and TRAAK, were expressed in Nav1.8+ Aβ-afferent TG neurons (Figure 4). In the TGs of Nav1.8ChR2/eYFP mice, eYFP was observed in many TG neurons, with their soma sizes ranging from small to large (Figure 4(a)–(c)). The large-sized eYFP-positive TG neurons (diameter > 34 μm, Figure 4) corresponded to nociceptive Aβ-afferent TG neurons defined by electrophysiology (see Figure 1), whereas the smaller eYFP-positive TG neurons were most likely to be nociceptive Aδ and C-afferent TG neurons (Figure 4(a)–(c)). The eYFP-positive TG neurons, including those of large size, exhibited strong immunoreactivity to TREK-1, TREK-2, and TRAAK (Figure 4(a)–(c), right panel). This indicates that temperature-sensitive K2P channels were highly expressed in nociceptive TG neurons, including nociceptive Aβ-afferent TG neurons.
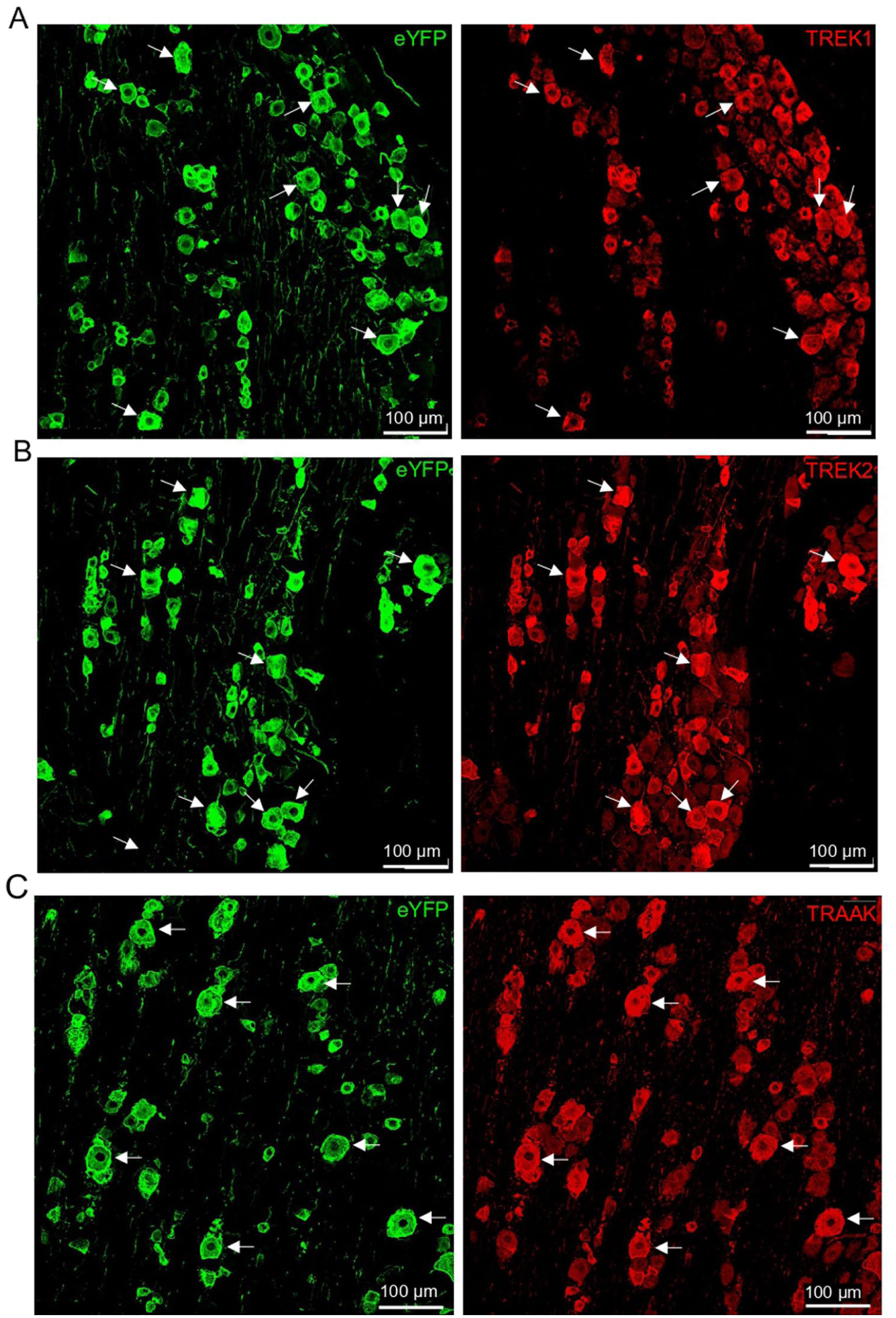
Expression of TREK-1, TREK-2, and TRAAK in Nav1.8+ Aβ-afferent TG neurons. Immunostaining in TG sections demonstrates the expression of temperature-sensitive K2P channels in Nav1.8+ (eYFP-positive) TG neurons. (a) Left panel: image shows immunostaining for eYFP expression in TG neurons. The eYFP-positive neurons were Nav1.8+ neurons. Arrows indicate some large-sized Nav1.8+ TG neurons (diameter > 35 μm), which correspond to Aβ-nociceptors. Right panel, same field as left panel, except that immunostaining for TREK-1 was shown. Arrows indicate large-sized TREK-1-positive TG neurons, which are also large-sized Nav1.8+ TG neurons shown in the right panel. (b) Similar to A except that eYFP (left panel) and TREK-2 (right panel) immunostaining was shown. (c) Similar to A except that eYFP (left panel) and TRAAK (right panel) immunostaining was shown. From A to C, Nav1.8+ TG neurons exhibit high expressions of TREK-1, TREK-2, and TRAAK. Scale bar: 100 µm.
We assessed the contribution of temperature-sensitive leak K+ channels to the total outward K+ currents in Nav1.8+ Aβ-afferent TG neurons. This set of experiments was conducted at 23°C in the absence and presence of norfluoxetine (250 μM) and Ba2+ (5 mM), two blockers for temperature-sensitive leak K+ channels. Outward K+ currents in response to depolarizing voltages were robust in Nav1.8+ Aβ-afferent TG neurons in the absence of the leak K+ channel blockers but substantially reduced in the presence of norfluoxetine plus Ba2+ (Figure 5(a) and (b)). The AUC of outward K+ currents was 386 ± 12 nA × mV (n = 8) in the absence of the leak K+ channel blockers but significantly reduced to 196 ± 14 nA × mV (n = 8) in the presence of norfluoxetine plus Ba2+ (Figure 5(c)). It should be noted that norfluoxetine and Ba2+ could block other K+ channels in addition to K2P channels, which prevent us from accurately quantifying the outward K+ currents that were only mediated by temperature-sensitive leak K+ channels.
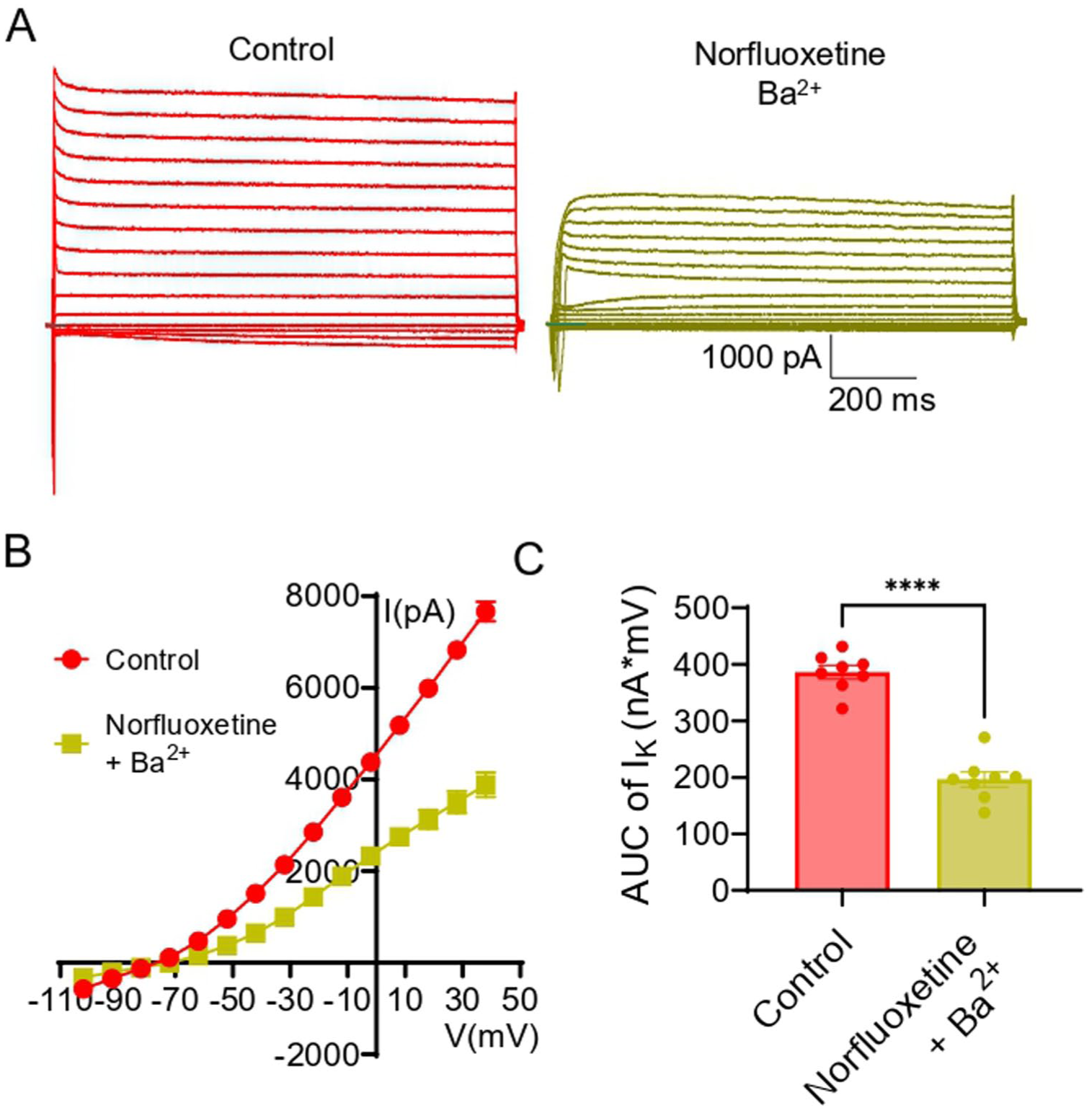
Pharmacological inhibition of K2P channels reduces outward K+ currents in Nav1.8+ Aβ-afferent TG neurons. Outward K+ currents were recorded in Nav1.8+ Aβ-afferent TG neurons under control conditions and following pharmacological inhibition of K2P channels using norfluoxetine (250 μM) and Ba2+ (5 mM). (a) Representative current traces show voltage-activated outward currents in control conditions and a marked reduction in current amplitude after application of the K2P channel blockers. (b) Current-voltage (I-V) relationships demonstrate a substantial decrease in outward currents across membrane potentials in the presence of norfluoxetine and Ba2+. (c) Quantification of outward K+ currents using the AUC of I-V plots reveals a significant reduction in currents following K2P channel inhibition. Data are presented as mean ± SEM, ****p < 0.0001.
To further characterize the effects of cooling on the leak K+ channels in Nav1.8+ Aβ-afferent TG neurons, we isolated leak K+ currents from the total outward currents by blocking voltage-gated potassium (Kv) channels with CsCl (135 mM), 4-AP (1 mM), and TEA (10 mM), and then assessed the effects of cooling temperatures on the leak K+ currents in Nav1.8+ Aβ-afferent TG neurons. We conducted this set of experiments at 23°C, 15°C, and 10°C, and omitted the test at 33°C (Figure 6) because there were no differences in the total outward currents between 33°C and 23°C (Figure 3(d)–(g)). In the presence of the Kv blockers and at 23°C, the remaining outward currents, that is, the leak K+ currents, were large (Figure 6(a)–(c)), over 40% of the total outward currents at 23°C (Figure 3(d)). The leak K+ currents exhibited temperature dependence, with a significant reduction at 15°C and 10°C compared to at 23°C (Figure 6(b) and (c)). Assessed by the AUC of I-V curves, the AUC was 175 ± 8 nA × mV (n = 10) at 23°C, significantly reduced to 96 ± 8 nA × mV (n = 9) at 15°C and 59 ± 8 nA × mV (n = 8) at 10°C in Nav1.8+ Aβ-afferent TG neurons (Figure 6(c)).
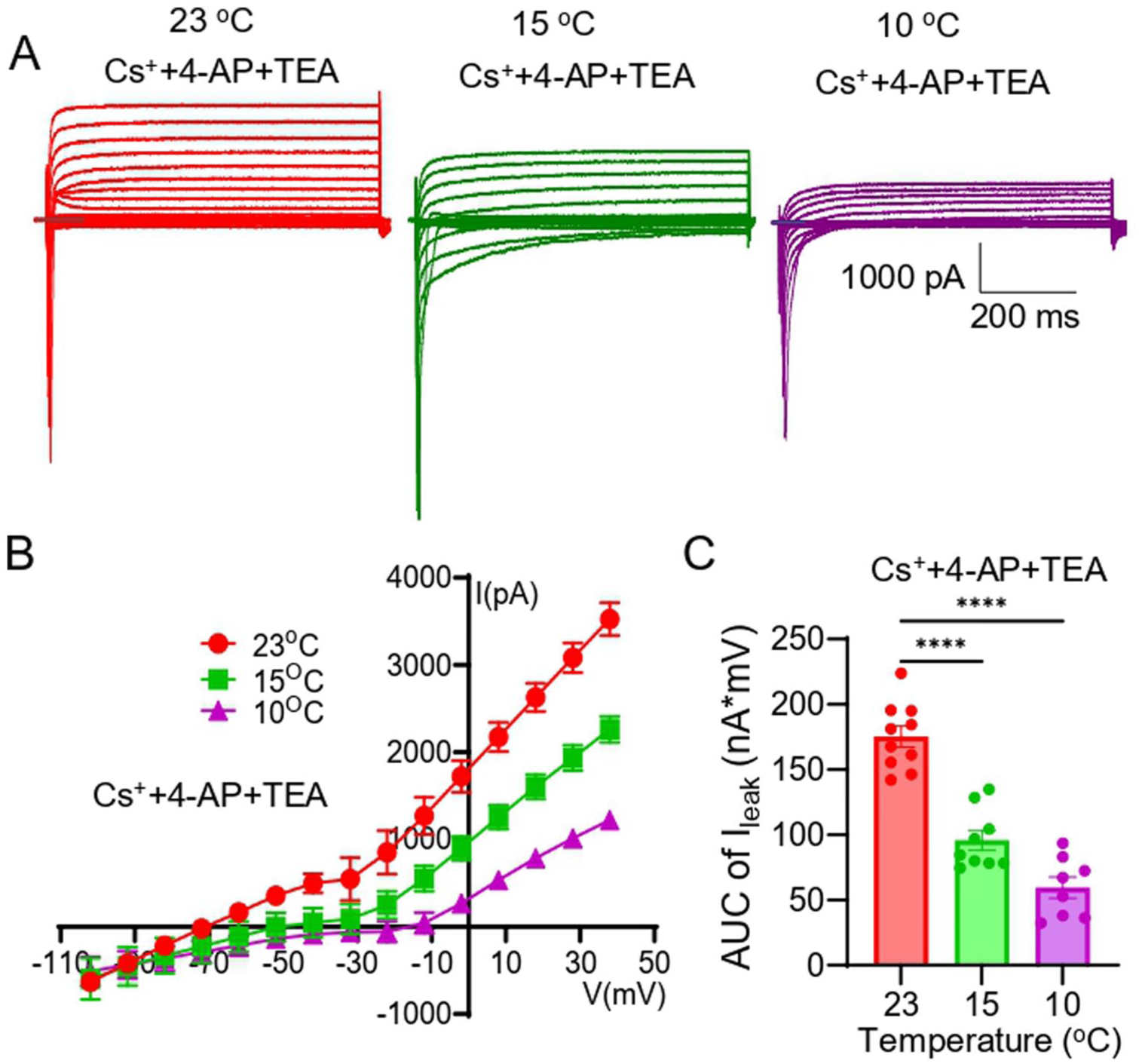
Cooling suppresses leak K+ currents in Nav1.8+ Aβ-afferent TG neurons. Leak K+ currents were isolated in Nav1.8+ Aβ-afferent TG neurons by blocking voltage-gated K+ channels using CsCl (135 mM), 4-AP (1 mM), and TEA (10 mM), and recordings were performed at 23°C, 15°C, and 10°C. (a) Representative traces of leak outward K+ currents recorded at 23°C (left), 15°C (middle), and 10°C (right), showing a progressive reduction in leak K+ current amplitude with cooling. (b) Current–voltage (I-V) relationships of leak K+ currents at 23°C, 15°C, and 10°C, demonstrating a temperature-dependent decrease in leak K+ currents across membrane potentials. (c) Quantification of leak K+ currents using the area under the curve (AUC) of I-V plots. Data are presented as mean ± SEM, ****p < 0.0001.
Discussion
In the present study, we have shown that cooling temperatures produce complex effects on the intrinsic membrane properties and excitability in trigeminal Aβ-afferent nociceptors, as indicated by recordings made from Nav1.8+ Aβ-afferent TG neurons in Nav1.8ChR2/eYFP mice. Cooling reduces conduction velocity and stimulation threshold in the axons of trigeminal Aβ-afferent nociceptors. However, cooling significantly increased the excitability in the soma of Aβ-afferent nociceptors. These effects are accompanied by cooling-induced suppression of activity on multiple ion channels, particularly temperature-sensitive K2P channels in trigeminal Aβ-afferent nociceptors. We have found that temperature-sensitive K2P channels, including TREK1, TREK2, and TRAAK, are highly expressed in trigeminal Aβ-afferent nociceptors. These findings highlight that temperature-sensitive K2P channels are key determinants of how cooling temperatures affect intrinsic membrane properties and excitability in trigeminal Aβ-afferent nociceptors.
Our results indicate that cooling enhances the excitability of the soma of trigeminal Aβ-afferent nociceptors, as evidenced by a reduction in AP rheobase, an increase in AP amplitude, an elevation of membrane input resistance, and a depolarization of resting membrane potential following cooling. A lower AP rheobase enables AP initiation by a smaller depolarizing current, thereby significantly contributing to the increased excitability.19,20 Increased membrane input resistance and a depolarized resting membrane potential may be responsible for the reduced AP rheobase. However, the reduced conduction velocity and increased stimulation threshold on the axons indicate that cooling temperatures also suppress the excitability of trigeminal Aβ-afferent nociceptors. Thus, cooling produces a dual effect: it enhances intrinsic membrane responsiveness on the soma while simultaneously limiting the efficiency of AP conduction on the axons of trigeminal Aβ-afferent nociceptors. Changes in intrinsic membrane properties and excitability in the soma may be attributed to the effects of cooling temperatures on the ion channel activity.14,21,22 Consistently, we have observed a reduction of inward currents mediated by voltage-gated Na+ channels at cooling temperatures. Although the inward Na+ currents are significantly inhibited by cooling, they remain large in amplitude even at 10°C. The inward Na+ currents may be largely mediated by Nav1.8 channels, since Aβ-afferent nociceptors highly express them. Previous studies have shown that Nav1.8 channels remain active, supporting action potential generation in nociceptors at low temperatures.9,22,23 Cooling temperatures are clinically used as a therapeutic modality for pain relief (e.g. cryotherapy). Our finding that cooling enhances the somatic excitability of these nociceptors seems paradoxical to their clinical analgesic effects. However, excitability will eventually decrease at colder temperatures due to more severe inhibition of voltage-gated Na+ channels. Although not recorded in this study, voltage-gated Na+ channels at the nodes of Ranvier of nociceptive Aβ-afferent fibers may also be suppressed at cooling temperatures, which may contribute to the decreased conduction velocity and increased stimulation threshold under cooling conditions.
In our study, potassium channels may play a key role in mediating the temperature-dependent effects on the intrinsic membrane properties and excitability of Aβ-afferent nociceptors. We have shown that cooling largely suppresses outward K+ currents in Aβ-afferent nociceptors. The outward K+ currents are partially mediated by voltage-gated K+ channels, as evidenced by the substantial inhibition of the outward K+ currents by voltage-gated K+ blockers. Importantly, the outward K+ currents are also largely mediated by K2P channels, as indicated by the inhibition of outward K+ currents by K2P inhibitors and the large remaining outward K+ currents in the presence of voltage-gated K+ blockers. We show that outward K+ currents mediated by K2P channels are highly temperature-sensitive and substantially inhibited by cooling temperatures in Aβ-afferent nociceptors. Consistently, we have observed high expression of TREK1, TREK2, and TRAAK, three temperature-sensitive K2P channels, in Aβ-afferent nociceptors. This cellular distribution supports the functional importance of temperature-sensitive K2P channels in intrinsic membrane properties and regulating excitability in Aβ-afferent nociceptors. The temperature-sensitive K2P channels provide a temperature-sensitive leak K+ conductance that is essential in controlling resting membrane potential and input resistance.16,17 Therefore, cooling-induced suppression of these K2P channels could depolarize the resting membrane potential and increase input resistance, leading to a lower AP rheobase in the soma of Aβ-afferent nociceptors. Furthermore, suppression of temperature-sensitive K2P channels at the nodes of Ranvier may account for the reduction of AP conduction on the axons of myelinated afferent nerves. 24 In addition to the cooling effects on temperature-sensitive K2P channels, the activity of voltage-gated K+ channels may also be partially suppressed by cooling, 22 although they are less temperature-sensitive than K2P channels. Voltage-gated K+ channels play a key role in AP repolarization, which determines AP width. 25 Suppression of voltage-gated K+ channel activity may be responsible for AP broadening at cooling temperatures.
Collectively, the molecular architecture of Aβ-afferent nociceptors, which combines Nav1.8 with temperature-sensitive K2P channels, underlies enhanced membrane excitability and the ability to generate APs at cooling temperatures. At the same time, altered conduction along the axons of these afferents may influence nociceptive signal processing at cooling temperatures. From a physiological perspective, changes in intrinsic membrane properties and excitability at low temperatures may represent an adaptive mechanism. Under progressively decreasing temperatures, Aβ-afferent nociceptors remain responsive, largely due to preserved Nav1.8 function and reduced K2P-mediated leak conductance. If these molecular arrangements occur in Aβ-afferent nociceptor endings, they may enable Aβ-afferent nociceptors to sustain nociceptive signaling to detect harmful stimuli with protective responses at low temperatures. Cooling-induced suppression of K2P channels and enhancement of the excitability of Aβ-afferent nociceptors may also have implications in neuropathic pain under pathological conditions.
Footnotes
Author contributions
JGG conceived the research project. JGG and ARS designed experiments. ARS performed electrophysiology experiments. JL created and maintained transgenic mice and performed immunostaining experiments. All authors participated in data analysis and/or interpretation. JGG and ARS wrote the paper.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by NIH grants DE018661 and DE023090 to J.G.G.