
Abstract
The term ‘N-of-1 trial’ has traditionally referred to a specific, well-defined, clinical trial methodology, involving evaluation of an intervention (or interventions) in a single individual, with alternating periods ‘on’ and ‘off’ an intervention. Given methodological constraints, such trials are best suited to the study of chronic, stable conditions, and so have had a limited role in progressive conditions like cancer. According to evidence-based medicine hierarchies, conventional, randomised, N-of-1 trials are considered ‘type 1’ evidence. In recent years, however, the term ‘N-of-1 trial’ has appeared with growing frequency within the academic cancer literature but with reference to a heterogeneous range of research activities (which diverge in various ways from the conventional N-of-1 trial design). In this qualitative study, 72 semi-structured interviews were conducted with key stakeholders across the cancer clinical trials landscape to explore perspectives regarding the notion of ‘N-of-1 trials’ in oncology. This included various types of ‘trial professionals’ (trialists, statisticians, ethicists and ethics review board members, industry, regulatory and reimbursement body representatives), as well as ‘consumer representatives’. Findings of this study highlight that two broad categories of issues are particularly salient to stakeholders in relation to single participant focused research: (1) issues to do with opportunity to participate, and (2) issues to do with consent and risk management. We draw attention to implications for those engaged in the review of various forms of single participant focused research, and how these differ from considerations pertinent to more traditional clinical research. In particular, we caution against any ‘one size fits all’ approach to assessment of ‘acceptable’ risk during ethical review of such activities. Finally, we consider whether novel forms of oversight may be required, including a possible role for ethics review boards to engage directly with individuals considering participation in such studies.
Keywords
Introduction
Systematic reviews of randomised controlled trials (RCTs) are conventionally positioned at the top of the evidence-based medicine (EBM) hierarchy (Centre for Evidence-Based Medicine, 2009). Methodological features such as randomisation and blinding mitigate the potential effects of bias and so increase the likelihood that the ‘true’ effect of a novel intervention may be estimated. Consequently, RCT-level evidence has traditionally been the mainstay for decisions about drug registration and reimbursement.
RCTs are, however, designed to estimate the aggregate effect of an intervention across a carefully selected (Kennedy-Martin et al., 2015) and managed trial population (Kyr et al., 2021). This creates difficulties both for translating efficacy in a trial setting to the effect in ‘real world’ (target) populations (the ‘efficacy-effectiveness’ gap (Templeton et al., 2020)), as well as for using population-level findings to guide the care of specific individuals (Kyr et al., 2021). In the modern era, increased appreciation of the role that differences amongst groups of individuals who may share the same disease but in reality represent highly heterogeneous populations adds a further tension to the conduct and interpretation of RCTs. In oncology, for example, advances in the molecular understanding of cancer have shed light on significant biological diversity between individuals who share the same cancer type (inter-individual heterogeneity), as well as ‘within’ individuals over the course of their illness (intra-individual heterogeneity 1 ).
In recognition of these challenges, some have called for a re-conceptualisation of the methods via which clinical evidence is generated and interpreted (Subbiah, 2023), including approaches to clinical trial design (Fountzilas et al., 2024; Fountzilas and Tsimberidou, 2018; Subbiah, 2023; Subbiah and Kurzrock, 2018). Novel research approaches which focus on the individual (or individuals) as the primary unit of investigation seek, arguably, to engage with the challenge that widespread clinical and molecular heterogeneity in cancer poses for other, more conventional, research methods. Heterogeneity is reframed from ‘statistical noise’ to ‘variability that carries information’, and so in need of accommodating as a source of knowledge (Kyr et al., 2021).
In some senses, there is nothing new about medical inquiry that centres the individual. Individual clinical experiences have been long documented in case reports and/or series and recognised for the role they can play in advancing understanding of conditions or treatments (Ankeny, 2020). However, such reports are typically observational and represent an attempt to explain some phenomenon of interest with the benefit of hindsight. This distinguishes case reports from ‘N-of-1 trials’, which are instead prospective, hypothesis-driven investigations in which a single individual 2 is subjected to alternating periods ‘on’ and ‘off’ an intervention (or series of interventions) (Mirza et al., 2017; Figure 1). When it comes to determining the effect of an intervention in a specific individual conventional, randomised, N-of-1 trials, which follow well-defined methodological standards (Porcino et al., 2020; Vohra et al., 2015), represent ‘type 1’ evidence in the EBM hierarchy (Chalmers et al., 2019; Guyatt et al., 1988). However, methodological requirements of the design constrain use for the most part to chronic, stable diseases. This is so that any change in the outcome of interest may be interpreted as due to the effect of the study intervention rather than (unrelated) evolution of the underlying disease (Hawksworth et al., 2024). Consequently, because cancer is usually a progressive disease application of the N-of-1 trial design in oncology has been restricted to studying interventions for symptom management rather than evaluation of systemic therapies aimed at tumour control.
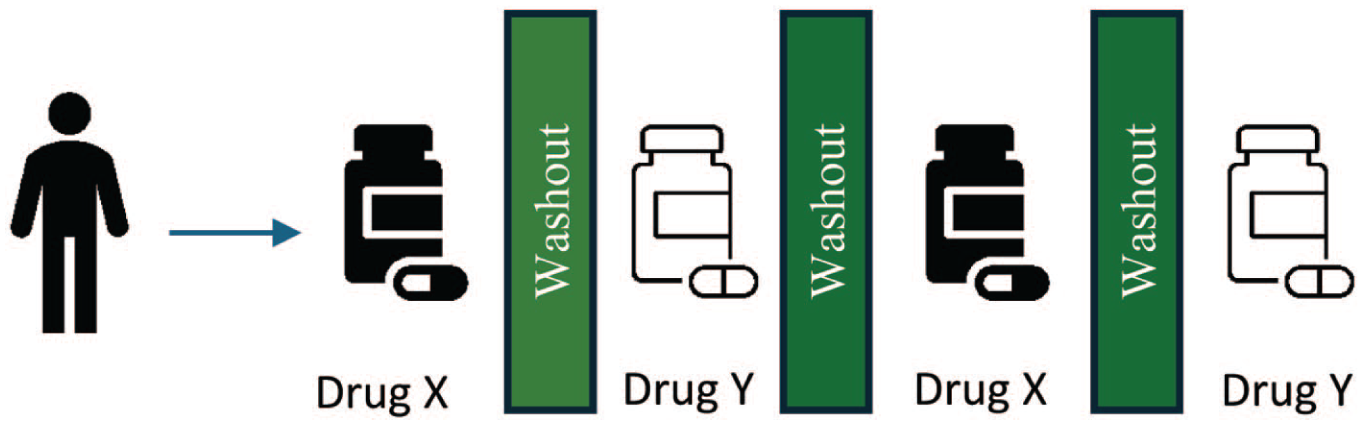
Conventional, randomised N-of-1 Trial. An individual is subjected to successive periods ‘on’ and ‘off’ a series of interventions. As the aim is to evaluate the effect of a study intervention on the outcome of interest within the individual, use of a conventional N-of-1 trial design assumes the underlying disease state remains similar (i.e. so any change observed can be interpreted as due to the study intervention).
Notwithstanding, recent and ongoing interest in research focusing on the individual has been buoyed by advances in understanding of the molecular basis and biological diversity of cancer (Fountzilas et al., 2024; Subbiah et al., 2024a). This has led to a notable increase in use of the label ‘N-of-1 trial’ in the academic cancer literature, however with reference to a heterogeneous range of research endeavours (i.e. characterised by a variety of methodologies, aims and phases of investigation; Collette and Tombal, 2015; Gouda et al., 2023; Kyr et al., 2021; Silvestris et al., 2017; Subbiah et al., 2025). Examples of the wide diversity of activities which have been referenced as ‘N-of-1 trials’ are outlined in Table 1. As we have described previously (Heynemann et al., 2025), this becomes problematic if epistemic claims to being ‘type 1’ EBM evidence are suggested (or assumed) of research activities beyond those which fit the methodological confines of conventional, randomised N-of-1 trials.
Use of the term ‘N-of-1 trial’ in oncology.

The design characteristics and implications of each of these types of studies have been described in more detail in an earlier publication (Heynemann et al., 2025).
That the label ‘N-of-1 trial’ has become such a slippery concept (Heynemann et al., 2025) has prompted Selker et al. to propose that conventionally defined (Porcino et al., 2020; Vohra et al., 2015), randomised N-of-1 trials be labelled ‘type 1 N-of-1 trials’ to distinguish them from other types of studies which diverge from this format (Selker et al., 2022, 2023). However, what they go on to propose as a ‘type 2 N-of-1 trial’ is also quite specific, being a ‘single-cycle pre-post design’, 3 and this proposal would fail to encompass the full spectrum of examples described in Table 1 (Selker et al., 2022, 2023). As such, in this paper we will use the term ‘N-of-1 trial’ only when referring to the conventional, randomised N-of-1 trial design. Otherwise, we will refer to ‘single participant focused research’ or ‘N-of-1 studies’, as ‘umbrella’ descriptors of the breadth of research activities referred to in the academic cancer literature which centre on the individual (or individuals).
Growing interest in various types of single participant focused research raises important epistemological questions such as: how should such entities be conceived in relation to more traditional forms of research, and do they even count as ‘research’ given they are not primarily aimed towards the production of generalisable 4 knowledge for populations (Kane et al., 2021). Tensions like these raise important questions about the ‘scientific’ and ‘social’ value of different approaches to research (Emanuel et al., 2000).
Single participant investigations also raise a range of ethical questions that could challenge concepts and processes usually relied upon by research ethics committees (RECs 5 ). For example, opportunity for withdrawal is usually considered an important component of consent to research participation. In single participant research, should the participant elect to withdraw this presumably would have greater impact on the ongoing viability of the study than in a traditional trial involving many participants. Given that ‘voluntariness’ is an important element of valid consent, this might require RECs to develop enhanced protections or novel frameworks for ensuring that consent remains truly voluntary throughout the research process. Separately, RECs might also need to reconsider some of their approaches to assessing and managing risk – or at least be alert to the highly varied risk profiles of different types of N-of-1 studies. These range from some of the highest risk studies (such as first-in-human dose-escalation studies per example (b), Table 1), to comparatively lesser-risk studies more akin to ‘innovative care’ 6 (i.e. utilising an already approved agent in a new combination per example (c), Table 1).
Whilst scholars have previously described, both conceptually (Crowden et al., 2015; Samuel et al., 2016; Stunnenberg et al., 2020) and empirically (Cen et al., 2016), ethical considerations pertinent to conventional, randomised N-of-1 trials, as noted, these diverge in various important ways from examples of single participant focused research described in the academic cancer literature. Moreover, for ‘type 1’ N-of-1 trials the primary moral concern has traditionally been centred on whether or not to categorise different examples as instances of ‘research’ or ‘care’ (Crowden et al., 2015). As we have argued previously, it is our contention that the types of single participant focused studies described to date in oncology are most accurately categorised as research, given each incorporate a prospective plan for learning and expose participants to various novel and uncertain elements (Heynemann et al., 2025).
To date, nobody has explored the perspectives of key stakeholders regarding these issues as well as potential ethical and other concerns about the types of single participant focused research being undertaken in cancer populations specifically. To address this gap, in this qualitative study we sought to explore the perspectives of researchers, ‘end users’ of trial results and consumer representatives regarding ‘N-of-1 trials’ in oncology.
Methods
Semi-structured interviews were conducted with individuals engaged in the conceptualisation, design, conduct, interpretation and translation of adult cancer trials as part of their professional role/s and consumer representatives advising on such activities. Individuals were identified for recruitment drawing on the collective experience of the author group in oncology, clinical trials and bioethics.
For Subgroup 1, herein the ‘trial professionals’ sample, individuals recognised as subject matter ‘experts’ or leaders in their respective disciplinary areas were approached. A variety of sampling strategies were used, including convenience (via existing networks of the author group), snowball (participant recommendations) and unsolicited email invitations to individuals identified via online profiles and publications. These strategies were used to promote maximum sample variation (e.g. by geography, tumour stream expertise, early-vs late-phase clinical trials focus, and statistical approach).
For Subgroup 2, herein the ‘consumer representative’ sample, individuals were recruited on the basis of experience acting in one or more advisory role/s related to cancer trials as a nominated ‘consumer representative’. Invitations were sent to ‘consumer advisory’ groups of trial co-operative groups, RECs involved in review of cancer trial protocols or individuals identified through professional contacts of the authors. These strategies were utilised to promote maximum variation in tumour streams represented (e.g. more vs less-common cancers), and types of consumer advisory input (e.g. processes related to conceptualisation, design, interpretation, ethical or other review of cancer trials).
Interviews were semi-structured in nature, to explore issues in depth whilst simultaneously facilitating some consistency of focus (interview schemas available as Supplemental Information ). In Subgroup 1, ‘N-of-1 trials’ were described to participants as trials in which a drug (or series of drugs) was evaluated in one individual, with the individual acting as their own control. For those who sought further clarification, description of a recent example of a study involving evaluation of a novel targeted therapy in single participants with a rare cancer subtype, which had been subsequently labelled as a ‘N-of-1 trial’ (Drilon et al., 2017) was also offered. Participants were also free to discuss other types of studies they considered to be ‘N-of-1 trials’. For Subgroup 2, diagrams of hypothetical ‘N-of-1 trials’ were used to assist discussion, assuming most consumer representative participants would have little awareness of the concept ( Supplemental Information ). These were presented as examples in which different agents were administered in an alternating pattern to a single individual, or in which increasing dosages of the same agent were administered.
Interviews were conducted by SH, a medical oncologist and experienced qualitative researcher, via ‘Zoom’, lasted between 25 and 80 minutes (Subgroup 1) and 22–69 minutes (Subgroup 2), and were audio-transcribed verbatim. Informed consent was obtained from all participants, either at time of completion of a brief demographics survey administered via REDCap or verbally prior to interview.
Findings reported here form part of a larger research project, ‘Exploring Stakeholders’ Views on Recent Trends in the Cancer Clinical Trials Landscape’, with ethics approval from The University of Sydney (2024/HE000229).
Results: Themes
Demographics
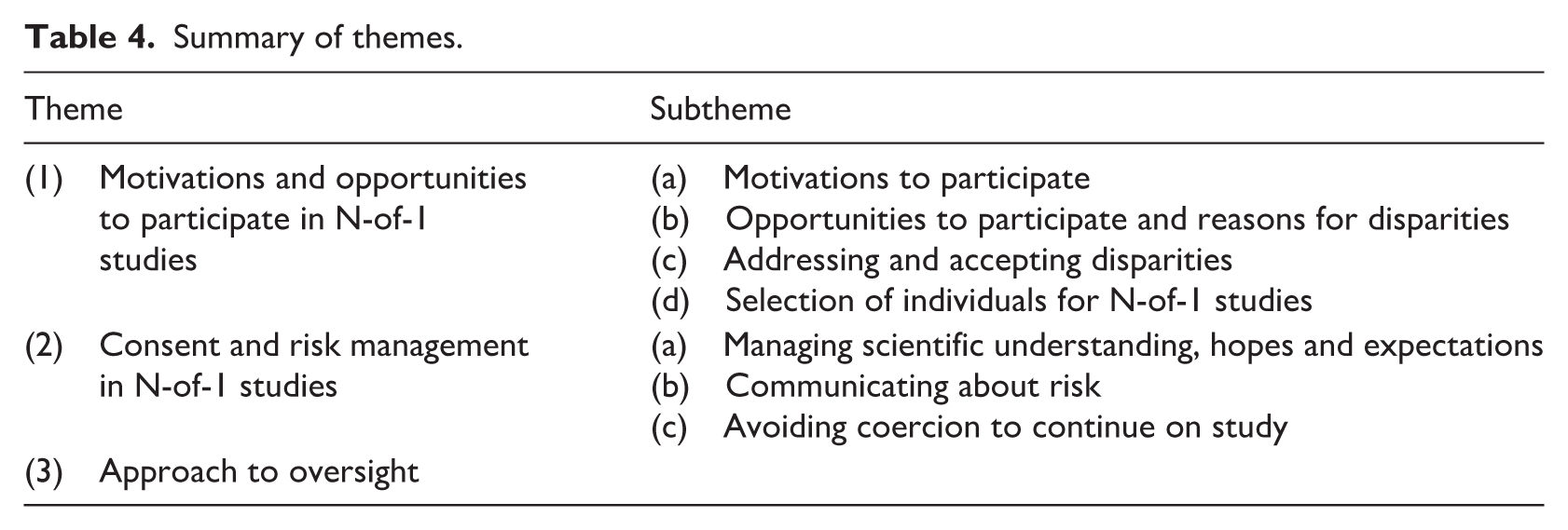
From May 2024 to 2025, 52 ‘trial professionals’ were interviewed, including: clinical trialists (CT, n = 20), regulatory and/or reimbursement body representatives (REG, n = 12), biostatisticians (STAT, n = 9), research ethics committee members and/or chairs (REC, n = 5), bioethicists (BE, n = 4), and industry representatives (IND, n = 2) with some individuals holding overlapping roles. From Sept 2024 to April 2025, 21 ‘consumer representatives’ were recruited with one lost to follow-up. For ‘consumer representatives’ we also denote tumour stream, and nature of lived experience represented (e.g. CONSUMERthoracic cancer, patient). Further details regarding participant demographics and expertise can be found in Tables 2 and 3, whilst Table 4 provides an overview of the thematic results.
Subgroup 1 ‘trial professionals’ demographic and expertise data (n = 52).
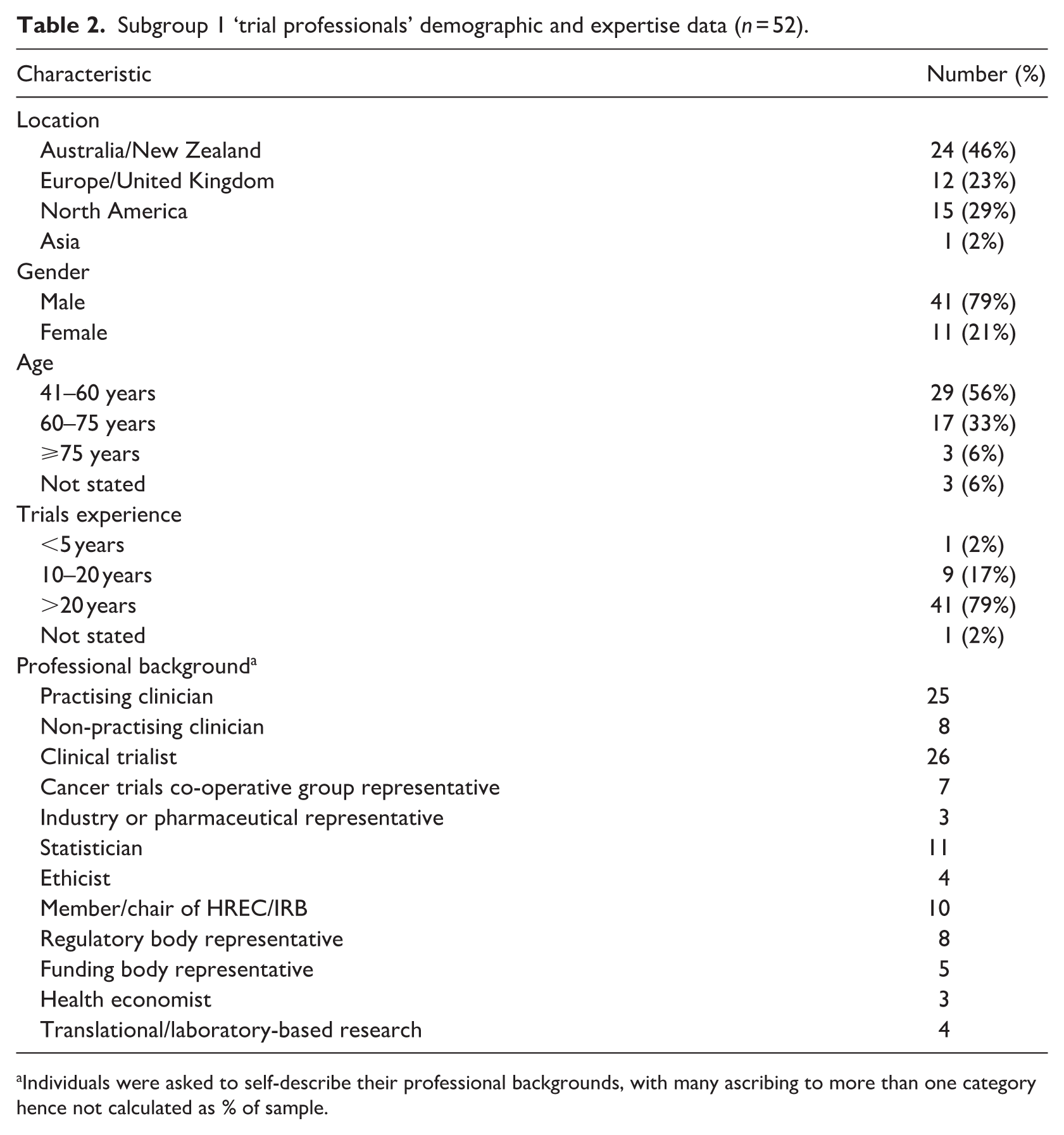
Individuals were asked to self-describe their professional backgrounds, with many ascribing to more than one category hence not calculated as % of sample.
Subgroup 2 ‘consumer representatives’ demographic and expertise data (n = 20 a ).

Twenty-one participants total were recruited and signed written consent to participate in the study, however 1 participant was lost to follow-up prior to interview completion.
Individuals were recruited on the basis of experience acting in one or more consumer advisory role/s, which included those on committees as a consequence of a ‘lived experience’ of cancer as well as those in other roles (e.g. REC or regulatory/reimbursement committee members).
Summary of themes.
Results
Theme 1: Motivations and opportunities to participate in N-of-1 studies
Subtheme 1a: Motivations to participate
Several consumer representative participants highlighted that altruism was an important consideration for some individuals in deciding to participate in clinical trials generally. Regarding N-of-1 studies specifically, several participants assumed they would not produce generalisable knowledge and so questioned whether involvement represented a worthwhile investment. One participant suggested that lack of generalisability was not just an ethical concern in relation to the participant, but also had societal implications in regard to resource allocation.
‘You’re doing it not just for your own survival. . .most participants see an element of altruism. . .if the outcome of that trial is only applicable to one person, and it’s not generalizable, I don’t think that’s just. . .I don’t think it’s. . .ethical. . . it’s not fair to use money and time for a trial that’s not generalizable to a greater good. . .I don’t think it’s fair for the society or whoever it is that’s funding this’. (CONSUMER01thoracic cancer, patient)
Trial professionals also recognised the limitations of N-of-1 studies with regard to the production of generalisable knowledge. As one participant noted ‘on the basis of one observation you can’t understand uncertainty’ (STAT02). Moreover, it was noted that for public decision-making ‘you still have to prove your drug at some point in a group because this is how decisions are made’ (REG12). However, only one participant expressed concern that limits to generalisability might affect consumers’ willingness to participate.
‘Even if you say to the patient, well, you’re participating in research that can inform the next patient who comes along. It’s not even clear how well that applies. . .the next patient who comes along is likely to get 3 different drugs from the ones that. . .Mrs. Jones is getting, and so how the information that Mrs. Jones is generating applies to Mr. Smith is also a bit unclear. . .’ (CT13)
The risk associated with being ‘first’ was another important consideration when it came to motivation to participate: ‘First-in-human, kind of raises eyebrows. . .there’s always a risk that something unforeseen may occur. . .If I was in a first-in-human trial, you know, it’d be nice to know I was the second guy rather than the very first guy’ (CONSUMER09paediatric neurological cancer, close contact, REC representative)
One participant considered participation in ‘N-of-1 trials’ as akin to being a ‘guinea pig’ (CONSUMER01thoracic cancer, patient), acknowledging that they found it difficult to imagine that someone with cancer would submit themselves to something like this.
‘To be able to do it you would need to give a lot of information about safety like really understanding that that person is going to be kept safe. And that there’s a lot of support. . .especially if we’re talking about cancer drugs. . .I actually can’t imagine being patient number one and one only’ (CONSUMER01thoracic cancer, patient)
The issue of being the only participant was also salient. Whilst it was recognised that involvement in trials with more participants did not necessarily guarantee safety, several participants considered it may be reassuring to know that more participants were being monitored.
‘It might all be a bit of a fallacy, but knowing that you’re 1 of 10 or 20 or 200 somehow conveys a sense of safety in numbers’ (CONSUMER08Breast Ductal Carcinoma In Situ (DCIS), patient, REC representative)
One participant even wondered whether more risk-taking might occur in the design of N-of-1 studies given only one participant’s life was at stake.
‘I don’t know if you’ve got a trial of one, whether you can be a little more, sort of ambitious, in the nature of the drugs that you use. . .you’re only playing Russian roulette with one person’s life as opposed to multiple people’s lives’ (CONSUMER16gastrointestinal cancer, patient)
Subtheme 1b: Opportunities to participate and reasons for disparities
Both trial professionals and consumer representatives were alert to the fact that recruitment to N-of-1 studies might not be even or equitable.
Several trial professionals argued that opportunity to participate was likely to vary widely based on a range of factors including the treating doctor’s knowledge, as well as the patient’s health literacy and self-advocacy: ‘All too often opportunities to participate are limited by either your knowledge as a patient or your clinician’s knowledge of what is available, and having access. Is that equitable? No, it’s not’ (REG02).
This was acknowledged to be a problem for trials access generally, but one that may be potentially intensified with N-of-1 studies. A trialist and practising clinician also wondered whether clinicians at more prominent hospitals, ‘ivory tower cancer centres’ or with closer ties to industry may be more likely to be involved in N-of-1 studies (CT14).
‘Maybe key opinion leaders that already have relationships with industry, for example, might get access. . .to these potential novel agents in an N=1 context more than other sites’ (CT14)
Conversely, several participants suggested that N-of-1 studies may be a mechanism for redressing inequities by serving as opportunities for individuals with rare cancers who were typically not eligible for ‘market share clinical trials’ (CT19). This was viewed as important given the perception that ‘every patient should, in theory, have access to at least 1 clinical trial. . .if they have a condition that is not curable with traditional treatment’ (CT19). Notwithstanding, such entities would be just the beginning of evidence generation in such settings, that ‘shouldn’t stop at N=1, that need to be part of the building block’ (CT03).
Separately, nuances of the local regulatory landscape were also anticipated to impact upon access to novel agents required for the conduct of N-of-1 studies. A non-US trialist considered the US to be a special context in which access to off-label drugs for individual patients was easier to procure. In contrast, the anticipated challenges of doing so in their own context posed a logistical barrier to conduct of N-of-1 studies.
‘In the US you can get a single patient, IND [investigational new drug], and [it’s] fairly easy to get that patient the drug. For us. . .either you pay for it through your own pocket for thousands a month, or somehow, if I write a protocol for it, and the [public health system] approves, and the company is willing to fund it through some sort of trial then I can do that. But most people don’t write protocols for different patients, separate protocols. It’s just too much work’ (CT12)
Subtheme 1c: Addressing or accepting disparities
Few suggestions were raised amongst the trial professionals as to how anticipated disparities in access to N-of-1 study opportunities might be addressed. A US-based trialist wondered whether compassionate access programmes, in which companies facilitate access to a novel agent might be a means of redressing possible disparities in access to N-of-1 studies due to geographic barriers.
‘I think that the compassionate use programs help with this. . .you have someone who’s geographically removed from, let’s say, an academic site where a trial is offered. They can’t travel there. And, you know, they can get access to a drug that’s not yet approved. . .I don’t know that it would be exactly the same as the. . .N-of-1 program I described’ (CT17)
Simultaneously, amongst the consumer representatives there was some evidence of acceptance of disparities in the service of longer-term goals. There was a sense that research had ‘to start with someone’ (CONSUMER14neurological cancer, close contact) and that sometimes this might include starting with just the one individual. This was not necessarily seen as problematic unless the agent had already been shown, or was shown quickly, to have real benefit.
‘If it’s a study drug and it’s early days, and they’re just trying to work it out, and they don’t know how well it’s going to work, doing it on one. . .you’ve got to start somewhere. . .the equity part only comes in if it’s. . .a bit of a hail Mary’ (CONSUMER09paediatric neurological cancer, close contact, REC representative)
There was also an assumption that study findings would be reported such that others could benefit from any learnings: ‘When you do an N-of-1 trial, you’re gonna write it up, right? So this is really a situation that could further. . .research. . .and really help other people’ (CONSUMER15thoracic cancer, patient).
Subtheme 1d: Selection of individuals for N-of-1 studies
Several consumer representative participants wondered how subject selection for N-of-1 studies would occur. For one participant, this was viewed as a potential area of concern if there were multiple individuals to choose between, wondering if a ‘drawing straws’ approach may be fairest, although they acknowledged that it would be difficult for those who drew the ‘short straw’ (CONSUMER16gastrointestinal cancer, patient).
‘So how do you select one out of presumably many?. . .if there’s potential for being, you know, quite a significant result it comes a little bit problematic. . .unless you’re the only one who has that particular cancer at that point in time . . .could be a bit of an ethical minefield’ (CONSUMER16 gastrointestinal cancer, patient)
In contrast, another participant challenged the idea of ‘exclusion’ in the context of N-of-1 studies. They argued that, while participants may be unfairly ‘excluded’ from other study designs, the very nature of the N-of-1 approach would necessitate selection of specific individuals.
‘It’s one thing not to include, it’s another thing to exclude. And so the other trials are more of the excluding, which is more of the equity piece, than this. . .the N-of-1 trial is so specific to a person’ (CONSUMER15thoracic cancer, patient)
However, the same participant also noted that it would be most likely that individuals selected for participation would be receiving treatment at an academic centre: ‘That’s where equity would come in, right?. . .it’s going to be the small percent of people that are treated at an academic centre’ (CONSUMER15 thoracic cancer, patient)
For trial professional participants, appropriate participant selection was raised more for the sake of data interpretation as well as risk management during an agent’s development, otherwise ‘you’ll kill it [the compound] before it gets started’ (CT02). Moreover, as a trialist and practising clinician suggested, there was also a sense that exposing unsuitable patients, in whom additional safety concerns might be anticipated, to these kinds of research would be unfair.
‘In an N=1 circumstance you’re not really gonna give something to someone who’s [got] really grossly deranged blood tests. . . you can’t make sense of the data. And again, on the principle of ‘do no harm’. . .if they [the patient has] got limited time and their time was going to be X. But now it’s going to be X minus something, because I’ve done this treatment to them then that’s not fair’ (CT02)
Theme 2: Consent and risk management in N-of-1 studies
Neither trial professionals nor consumer representatives saw consent requirements for N-of-1 studies as being unique. As one consumer representative explained, general research ethics guidelines should be applied, with the option of adding specific considerations related to risk or as raised by the ethics committee, following review of a study.
‘There’s a template right, which is your basic benchmark. . .after that, if there’s other things that have been raised by way of ethics, or by way of complexity or, you know, an incredibly sort of experimental nature that does need some more sort of caution then you add on, as opposed to create something new, because principles are principles’ (CONSUMER04consumer representative on regulatory/reimbursement body)
Despite denying (or, in the case of trial professionals, not raising) the need to fundamentally reconceptualise consent, participants in both subgroups emphasised the need to attend to a range of issues that are particularly salient or acute in this context.
Subtheme 2a: Managing scientific understanding, hopes and expectations
Several consumer representative participants argued that it would be important for individuals to know if they would be the only participant in an N-of-1 study, and cautioned that this might be difficult for individuals to understand.
‘I think it’s really hard, ethically like. . .I think people have a right to know that they are, that this is an N-of-1 trial, and why? . . . I mean, in what circumstances do we just want one participant as opposed to a small group’ (CONSUMER01thoracic cancer, patient)
Another participant worried that, in the absence of any such explanation and justification, individuals might mistakenly develop the impression that investigators ‘don’t know what you’re doing, even though that’s not the case’ (CONSUMER02gastrointestinal cancer, patient).
While consumer representatives were most concerned about scientific misunderstandings, trial professionals were more concerned with managing participants’ hopes and expectations.
This was seen to apply to all forms of single participant focused research however noted to be a greater potential concern in those undertaken in early-phase settings. Here, the single participant nature of the study was seen to exacerbate well-known challenges of managing expectations in early-phase studies when individuals have few or no therapeutic options, and so might view study participation as their only way of accessing ‘treatment’.
‘It’s already a lot of managing expectations. . .those expectations are potentially even more intensified when it’s a single person having a medicine’ (CT14)
A tension was, however, evident here. On the one hand, patients who believed they would benefit were seen to harbour a therapeutic misconception of personalised care, which needed ‘managing’. Simultaneously, it was acknowledged (somewhat ambivalently) that such trials are often intended to benefit individuals as well as generate evidence. As a trialist described, N-of-1 studies are effectively a ‘self-fulfilling prophecy’, given that their primary goal is ‘to try and treat a particular patient in a particular way, and the patient is, I’m assuming, part of that sort of contract’ (CT03).
Another trialist made a similar point: ‘Clinicians are running these trials hoping to see clinical benefit. I don’t know of any oncologist. . .that runs trials just to get data’ (CT19)
One consumer representative participant, who themselves had asked their treating physician if they could be an ‘N-of-1 patient for research’, affirmed this sentiment, conceptualising such opportunities as the ‘epitome of personalised medicine’ (CONSUMER15thoracic cancer, patient).
An ethicist was explicit about this tension surrounding management of participant expectations, conceptualising N-of-1 studies as representing a liminal space in the increasingly seamless research-care interface in oncology. That is, whilst the focus was treatment for an individual, this was being undertaken based on less evidence than would be expected for ‘routine’ practice—that is, as ‘experimental care’ (BE02).
‘It’s not just two scenarios, clinical care and research. . .rather there’s the middle scenario, which is providing clinical care, but non-standard clinical care, and I suspect that’s an important component of the N-of-1 trial because you may be doing stuff to this patient that’s non-standard but you’re not sure whether it’s gonna work or not so you want the consent to be clear in lay terms on what it means to be providing treatment that is untested and you’re not sure whether it’s gonna work or not. . .’ (BE02)
The ambiguity between research and non-standard care was further complicated by practical considerations regarding the regulatory and reimbursement status of interventions used in N-of-1 studies, and extended to questions about who should bear the costs of unproven interventions.
‘The drugs in most cases are going to be all off-label or some combination of off-label and investigational. . .that also introduces a real ethical dilemma in terms of. . .can you ask patients to spend what would likely be tens of thousands of dollars a month out-of-pocket for off-label treatment of unproven efficacy’ (CT13)
Subtheme 2b: Communicating about risk
Participants in both sub-groups argued that, in some instances, N-of-1 studies may pose a high level of risk and emphasised the need to ensure that participants understood this.
At what point use of an N-of-1 approach might be most appropriate in the developmental trajectory of an agent was conceived variably amongst several trialists, with implications for both evaluation of safety and methodological reasons. As one trialist observed, ‘The drug’s gotta get safety upfront before you can start applying it. So I’m thinking that you’re in a Phase 1b situation [dose-expansion], not a Phase 1a [dose-escalation]’ (CT02)
Conversely, a different trialist described their experience conducting single-participant dose-escalation studies, highlighting the importance that N-of-1 studies in this setting probably represented the riskiest form of research, and communicating this to participants was key.
‘We highlighted things like this has never been given to a human being before. . .however, there’s very strong data that provides a rationale for use. We don’t know what the side effects are. However, we think that, based on the class of drugs, they could be A, B and C. So fundamentally, I don’t think the approach to informed consent should differ. It should really focus on. . .it is a higher risk, it’s arguably the highest risk, one of the highest risk protocols, and that should really come out in the process’ (CT17)
Consumer representative participants emphasised the importance of informing participants about the study phase and, in particular, whether the study was a first-in-human evaluation and/or involved use of unapproved agents. One participant (who assumed that such studies would occur in the first-in-human setting) suggested that the opportunity to participate should be introduced as an alternative to symptom-directed supportive care, and that participants should be made aware that they could end up ‘worse off’.
‘Coming back to consent. . .you’ve been unresponsive to other treatments. And you have a choice of palliative care, or to try this. . .You probably want to give that sort of information as to. . .is it a new treatment? Is it something that was tried before and it looked like it might be promising and we want to get some more data. . .we don’t really know - it may actually make you worse off’ (CONSUMER18genitourinary cancer, patient)
Another consumer representative participant suggested that individuals should be made aware that participation in this kind of study may preclude them from participating in other potential trials.
Despite concerns expressed about the safety of N-of-1 studies, several consumer representative participants noted that receptivity to risk/s associated with research participation varied between individuals and that ultimately it was for each (informed) person to decide what was acceptable to them. It was also recognised that individuals with limited standard therapeutic options might be more likely to participate in trials with intensive requirements, or high levels of uncertainty including regarding possible risks. As described by a consumer representative on a REC, ‘These people have been through everything. . .it’s the roll-of-the-dice time. . .’ (CONSUMER09paediatric neurological cancer, close contact, REC representative)
Trial professionals noted, similarly, that it was ultimately up to patients to decide how much risk they were willing to take on. One trialist noted that, in essence, individuals were being offered the opportunity to be ‘their own experiment’ (CT16) which may be attractive for some but likely not all patients. Ultimately, they suggested that the decision to participate in such studies would be subject to the individual believing that the doctor had their ‘best interests at heart’, felt it was a ‘reasonable thing for them’, and the individual’s family agreed with this appraisal (CT16).
Subtheme 2c: Avoiding coercion to continue on study
One consumer representative participant noted that being the sole participant of a study may create psychological pressure to continue on study, particularly if the participant had good rapport with the investigator. Consequently, it would be important to ensure that the option of study withdrawal was underlined during the consent process.
‘Being part of something where it all hinges on you, it occurs to me that the pressure that you might feel once you decide to even think about doing it would be quite different than if you knew just 1 of 10 or 200, where the dropout rate, it won’t matter. . . .especially if there’s urgency to test this thing or drug. . .So dealing with that. . .in a way that. . .really convinces the participant that they can withdraw would be important’ (CONSUMER08 Breast Ductal Carcinoma In Situ (DCIS), patient, REC representative)
Theme 3: Approach to oversight
Views differed regarding the governance of N-of-1 studies. For some participants, having some form of oversight was an important safeguard.
‘The usual trial process. . .some sort of peer review of process. . .that there’s people outside looking in saying is this safe what you’re doing. . .measure things along the way, if it’s unsafe. . .pull out. You can imagine that wouldn’t happen if there wasn’t that oversight in an N=1 trial. If it’s just the investigator and the patient’ (CT14)
After all, as a trialist and proponent of N-of-1 studies pointed out, ‘even if in a N-of-1 study, not all studies are going to be positive’ (CT06).
Conversely, another participant who was a member of a REC, and conceptualised the N-of-1 concept as more closely resembling a care-related activity, did not feel review by a research-focused ethics committee would be necessary.
‘I wonder if it should actually even go to a REC. Because is it research? . . .I think RECs can get a bit carried away. . .they’re not treatment ethics committees. . .we’d often to be asked to comment on something, and I’d say, no, no, that’s a treatment ethics issue. So here, with an N-of-1, to me, that’s between the treating doctor and the patient. And my understanding is, you can do whatever you want, actually, as long as the patient consents, as long as they have an explanation’ (REC03)
Discussion
Growth in interest in single participant focused research in oncology raises important ethical questions regarding the justification, conduct and oversight of such activities. Our study has shown that two broad categories of issues are particularly salient to stakeholders, those to do with (1) opportunity to participate and (2) consent and risk management.
Fair opportunity to participate in N-of-1 studies
Fairness in subject selection, as well as equity of participation opportunities are both recognised as important elements of ethical research conduct (Emanuel et al., 2000). These ideas are underpinned by justice-related principles, such that the potential benefits and burdens of research participation are evenly distributed across populations.
Disparities exist across all domains of research, including the cancer trials landscape, with respect to under-representation of particular demographic groups (e.g. culturally and linguistically diverse populations (Muhandiramge et al., 2025; Pittell et al., 2023), lower socioeconomic status (Guadamuz et al., 2024), older individuals (Nguyen et al., 2025; Singh et al., 2017)) in addition to well-described structural barriers to trial access and participation (geographic location (Kirkwood et al., 2025), financial barriers (Williams et al., 2024)). Various suggestions have been proposed, at both local and international levels, to address the unique and overarching themes in each of these domains. These include promotion of trial consortia (to facilitate collaboration, awareness, and potential for cross-referrals between sites; Liu et al., 2025), moves towards de-centralised or ‘tele-trial’ networks (Underhill et al., 2024), initiatives to improve availability of culturally-sensitive and language-specific trial materials (Pal et al., 2023), amongst many others.
These general disparities are likely to manifest in similar ways in N-of-1 studies, and in many cases be managed in similar ways. Our participants did, however, make some observations that might require special consideration in the N-of-1 context.
The first is that access to N-of-1 study opportunities is likely to be shaped by factors related to an individual’s treating clinician, and so a potential source of inequity in regard to the likelihood of an individual being offered such opportunities. Awareness of study opportunities is, of course, a well-known issue affecting trial participation (Unger et al., 2021), with trials (particularly early-phase ones) conducted disproportionately in academic centres. However, even in these cases, there are usually multiple clinicians involved across a range of institutions. In the N-of-1 context, opportunity or otherwise for individuals to participate is likely to be more heavily influenced by factors related to a patient’s treating clinician – including their knowledge and/or awareness of potential N-of-1 study opportunities, as well as attitudes towards perceived risks versus benefit/s of such endeavours. In this sense, access to N-of-1 studies may introduce similar challenges to those identified in relation to access to novel agents via accelerated access programmes, in which different clinicians act as either ‘gatekeepers’ or ‘guides’ towards such opportunities (Pace et al., 2021).
Interestingly, concerns regarding fairness and equity in accessing N-of-1 study opportunities appeared to be somewhat attenuated, or at least conceived differently, amongst the ‘consumer representative’ sample. While consumer participants saw it as problematic if there was uneven opportunity to access highly effective agents, they recognised that research must ‘start somewhere’ and that individuals electing to participate in N-of-1 studies were still taking on some risk to themselves in doing so. Unless deliberate exclusion occurred, it was seen as reasonable to target involvement of specific individuals initially. In this regard, there might be some interesting (actual or perceived) differences between the attitudes of consumers towards the opportunity to participate in N-of-1 studies versus other, traditional trial opportunities. Not being offered participation in traditional trials may be seen as a genuine ‘missed opportunity’ whereas, at least for some, this may be less concerning for N-of-1 studies if conceived more as preparation for ongoing investigation.
Another interesting finding pertained to the implicit distinction that consumer participants made between choosing participants purposively because of their biology (which was acceptable) and ‘drawing straws’ (which was not). This finding suggests that the degree to which someone understands the custom designed nature of N-of-1 studies, in response to the clinical and biological characteristics of a specific individual’s cancer, is likely to have a bearing on the perceived fairness of subject selection procedures.
Consent and risk management
Informed consent has long been recognised as an important component of ethical research conduct. This is based on recognition of the importance of respect for autonomy, noting that ultimately it is the prerogative of each individual to determine if participation in a particular research activity coheres with their ‘best interests’ (Emanuel et al., 2000).
The importance of risk management in clinical research is well outlined in the research ethics literature. This is often woven into discussions of consent, with the idea being that if an individual makes an informed and non-coerced decision to participate in a study, they have presumably come to a determination that both the potential benefits and risks of participation are acceptable to them (both the magnitude and nature of possible risk/s and benefit/s, as well as relative to other opportunities).
For all but the most libertarian bioethicists, however, risk management does not collapse completely into consent, not least due to the range of factors recognised as potential obstacles to achieving genuine informed consent (e.g. health literacy, information ‘overwhelm’ (Bester et al., 2016)). Consequently, ethical justification of exposure to risk in clinical research is also predicated on the impression that a proposed research activity is characterised by a more objectively ‘favourable’ risk-benefit ratio as determined by a REC (Emanuel et al., 2000). That is, someone other than the patient makes a determination that ‘risks to the subject are proportionate to the benefits to the subject and society’ (Emanuel et al., 2000). In some ways, there is nothing different about consent and risk management in N-of-1 studies, but our participants did point to some issues that might require special consideration.
Ensuring adequate understanding and communication about risk
First, while risk is always assessed on a case-by-case basis by RECs, in the N-of-1 context it will be especially important to recognise the diversity of activities encompassed by the single label ‘N-of-1’. Importantly, these range from some of the ‘riskiest’ (CT17) possible research that an individual could choose to involve themselves in to studies much less likely to make participants ‘worse off’.
In this regard, it is noteworthy that some of our participants seemed to implicitly equate N-of-1 studies with highly risky first-in-human studies, whereas others seemed to equate N-of-1 studies with ‘personalised care’. Both assumptions are potentially correct in some cases and significantly misguided in others, and both types of potential misperceptions would need to be recognised and addressed in managing consent for different types of N-of-1 studies.
Ensuring adequate understanding and communication about benefit
There has been discussion in many contexts about the degree to which clinical trials (also) represent therapy (Burris, 2020; Kimmelman, 2020; Shalowitz and Miller, 2024; Subbiah and Kurzrock, 2025; Wilson et al., 2024), and whether individuals who assume they will receive therapeutic benefit from trial participation harbour a ‘therapeutic misconception’ (Appelbaum et al., 1982). Elsewhere, we have argued that traditional understandings of the ‘therapeutic misconception’, which draws attention to situations when trial participants conflate research with care, is a problematic conceptualisation in the increasingly enmeshed research-care interface in oncology (Heynemann et al., 2024).
We have also argued that the types of N-of-1 studies reported to date in oncology should be categorised, primarily, as ‘research’ endeavours warranting REC oversight (Heynemann et al., 2025). However, as our study findings highlight, it is simultaneously important to acknowledge that when opportunities to participate in N-of-1 studies are pursued by clinicians and patients they are also conceived, inextricably, as opportunities for ‘care’ (albeit far from the ‘routine care’ sense). For this reason, it will be important for RECs to (1) consider that both the research and care ‘hybrid’ (Johnston et al., 2025) nature of N-of-1 studies is portrayed fairly in patient information materials, as well as (2) be cautious to avoid undue paternalism when making determinations of ‘acceptable’ risk in N-of-1 study proposals (Edwards et al., 2004; i.e. given individuals confronted by difficult circumstances may well have an altered risk appetite (Gaskin et al., 2004)).
A further challenge for informed consent relates to the degree to which individuals understand – and are motivated by – the possibility of contributing to advances in scientific understanding via participation in research. On the one hand, as our participants demonstrated, there may be a (false) assumption that N-of-1 studies contribute no social benefit whatsoever. However, whilst N-of-1 studies will likely differ in their capacity to contribute to the advancement of knowledge in broadly generalisable ways, they may still advance learning in a ‘locally generalisable’ manner (Heynemann et al., 2025). Notwithstanding, it will be important that RECs ensure participants in N-of-1 studies are appraised of the epistemic potential of such endeavours, without either over- or under-stating this. Failure to recognise and communicate this might represent and exacerbate what Earl et al have recently labelled the ‘social value misconception’ – a possible misunderstanding amongst research participants about the ‘potential benefits of research for other people’ (Earl et al., 2025). An exaggerated assumption regarding social benefit of any research (including, but not limited to N-of-1 studies) may be problematic if an individual decides to participate on the basis of ‘false beliefs’ regarding the nature and/or likelihood of a study’s scientific contribution.
Suggestions for oversight
Whilst as noted earlier, the types of N-of-1 studies described in oncology cohorts to date all warrant formal REC review, we note that there are some unique features of N-of-1 studies that might necessitate bringing in other forms of oversight. As highlighted in our study, there are arguably unique tensions surrounding the ‘voluntariness’ of participation in N-of-1 studies in oncology related to study withdrawal. While it may be challenging for participants to withdraw from any kind of study, it could be particularly difficult to withdraw from N-of-1 studies if they are the only participant and feel obliged to continue for the sake of the research generally, or out of loyalty to the investigator who may also be their treating clinician.
For these reasons, there might be a role for RECs to engage directly with prospective trial participants, particularly when it comes to the ‘riskiest’ (CT17) forms of N-of-1 research proposals. 7 Alternatively, there might be a role for an independent clinician (or clinician-ethicist, if available) with expertise in the relevant disease area to be involved in REC determinations. We suggest that such an individual could play two specific roles: (1) to ensure any relevant potential conflicts of interest or commercialisation implications are made transparent (Heynemann et al., 2025), and (2) to explicitly reiterate the opportunity to withdraw participation, noting this is likely to be more challenging. The precise roles and responsibilities of such advocates would need to be carefully defined – for example, they could not be expected to audit participant understanding or predict likely regret.
Study limitations
As with all qualitative research, our findings are context specific and reflect the perspectives of participants included. Whilst a relatively large sample size for a qualitative study, it is notable that many of our participants were based in Australia, and a few specific stakeholder groups proved challenging to recruit and so represent a gap in the perspectives captured (e.g. for subgroup 1,industry and US-based regulatory representatives). For subgroup 2 specifically, whilst recruitment aimed to promote diversity amongst tumour types represented (e.g. more vs less common cancers), diversity in other domains was limited by issues in ‘representativeness’ of individuals involved in consumer representative roles more generally (e.g. lack of diversity in educational levels, geography, ethnicity (Kiss et al., 2024)).
It is also possible that our sampling approach may have led to ‘particular’ types of participants being recruited – such as those able to allocate time to be interviewed or who held strong views about ethical issues pertaining to cancer trials. That we observed a diversity of opinion across a range of topics somewhat attenuates this concern, though comparison of our study findings in future with other methods such as focus groups and surveys would be of interest. Additionally, whilst in this study we did not capture participant ethnicity, noting broader challenges relating to lack of diversity amongst those engaged in cancer trials from design and conduct as well as participation, attention to exploring perspectives of typically under-represented groups would also be of interest. Finally, in this study we sought to explore stakeholder perspectives regarding ‘N-of-1 trials’ in oncology broadly, however there is merit in further exploration beyond oncology.
Conclusion
While single participant focused research activities remain uncommon in oncology, it is important that those engaged in the ethical oversight of cancer research be aware of their existence and attendant ethical implications. Such activities are not the same as conventional, randomised N-of-1 trials conducted in other, non-cancer settings, and they also differ markedly from traditional cancer trials involving groups of participants. Consequently, usual approaches to ethical oversight of these kinds of research cannot merely be transposed to review of N-of-1 studies in oncology.
Supplemental Material
sj-docx-1-rea-10.1177_17470161261463012 – Supplemental material for Stakeholder perspectives regarding single participant research in oncology
Supplemental material, sj-docx-1-rea-10.1177_17470161261463012 for Stakeholder perspectives regarding single participant research in oncology by Sarah Heynemann, Wendy Lipworth, Sue-Anne McLachlan, Jennifer Philip, Tom John and Ian Kerridge in Research Ethics
Supplemental Material
sj-docx-2-rea-10.1177_17470161261463012 – Supplemental material for Stakeholder perspectives regarding single participant research in oncology
Supplemental material, sj-docx-2-rea-10.1177_17470161261463012 for Stakeholder perspectives regarding single participant research in oncology by Sarah Heynemann, Wendy Lipworth, Sue-Anne McLachlan, Jennifer Philip, Tom John and Ian Kerridge in Research Ethics
Supplemental Material
sj-docx-3-rea-10.1177_17470161261463012 – Supplemental material for Stakeholder perspectives regarding single participant research in oncology
Supplemental material, sj-docx-3-rea-10.1177_17470161261463012 for Stakeholder perspectives regarding single participant research in oncology by Sarah Heynemann, Wendy Lipworth, Sue-Anne McLachlan, Jennifer Philip, Tom John and Ian Kerridge in Research Ethics
Supplemental Material
sj-docx-4-rea-10.1177_17470161261463012 – Supplemental material for Stakeholder perspectives regarding single participant research in oncology
Supplemental material, sj-docx-4-rea-10.1177_17470161261463012 for Stakeholder perspectives regarding single participant research in oncology by Sarah Heynemann, Wendy Lipworth, Sue-Anne McLachlan, Jennifer Philip, Tom John and Ian Kerridge in Research Ethics
Footnotes
Ethical considerations
Ethics approval for this study ‘Exploring Stakeholders’ Views on Recent Trends in the Cancer Clinical Trials Landscape’ was granted from The University of Sydney (2024/HE000229).
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Sarah Heynemann has been supported by the National Health and Medical Research Council (NHMRC) postgraduate scholarship scheme (2022/GNT2021953).
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tom John: Honoraria/Advisory (BMS, AstraZeneca, Amgen, Arivent, Roche, Pfizer, Takeda, Boehringer Ingelheim, MSD, Merck, Puma, Specialised Therapeutics, Gilead, Seagen, Johnson and Johnson, Bayer, Beigene), Travel/speaker fees (AstraZeneca, Beigene, Dizal, MSD). Sue-Anne McLachlan: Honoraria/Advisory (BMS). Sarah Heynemann: Travel fees (AstraZeneca). Jennifer Philip: Speaker fees (Roche). Wendy Lipworth/Ian Kerridge: Nil.
Supplemental material
Supplemental material for this article is available online.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.