
Abstract
Cerebral autoregulation is the fundamental mechanism that regulates cerebral blood flow in the face of blood pressure variability. It is a dynamic process that depends on vascular smooth muscle cell contractile function, driven by myogenic, chemical, and autonomic mechanisms. Cerebral small vessel disease (cSVD) is associated with increased blood pressure variation due to systemic hemodynamic dysfunction, as well as dysfunction of the cerebral small vessels, characterized by endothelial dysfunction and vascular reactivity. As such, cerebral autoregulation may be impaired in cSVD, either as a manifestation of the disease or as a cause. This narrative review addresses current clinical evidence for involvement of autoregulatory dysfunction in cSVD. It discusses the mechanisms of autoregulation; how these overlap with vascular mechanisms involved in cSVD; methods of measurement of autoregulation in these patients; evidence for altered autoregulation in patients with either sporadic cSVD, previous stroke, or specific forms of cSVD; and effects of potential therapies for cSVD on autoregulation. This review thereby identifies the potential importance of autoregulation in cSVD, highlights current limitations in our understanding, and outlines future research avenues.
Keywords
Introduction
Cerebral small vessel disease (cSVD) affects the structure and function of the small blood vessels in the brain. It is an ubiquitous feature of brain imaging with aging, 1 rising from 50% prevalence by age 60 to greater than 80% by 80, 2 but despite this, it remains a largely unrecognized public health challenge. cSVD results in ~30% of ischemic strokes, 3 80% of hemorrhagic strokes, 4 and 40% of dementia and is a key cause of late-onset refractory depression, 5 apathy, and impaired mobility. 6 It is, thus, not a feature of aging, but a pathology of aging.
The heterogeneity of cSVD limits our mechanistic understanding. cSVD is an umbrella term for chronic damage to small cerebral blood vessels resulting from multiple pathologies, ranging from monogenic disorders (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL, Col4A1/A1) 7 and hypertension-associated deep perforator arteriopathy, 8 to distinct molecular mechanisms such as vascular amyloid deposition in cerebral amyloid angiopathy. 9 Sporadic cSVD is principally defined on MRI by white matter hyperintensities, microbleeds, dilated perivascular spaces, and lacunar strokes, as well as atrophy, cortical microinfarcts, microstructural damage, and convexity superficial siderosis. 1 However, evidence relating each of these features to underlying mechanisms is limited.
Recent research is beginning to unravel this complexity through measuring interactions between transmission of dysregulated systemic hemodynamics (hypertension, aortic stiffening, arterial pulsatility) and dysfunctional compensatory cerebral hemodynamics, 8 regulated by local endothelial cell dysfunction; their regulation of vascular smooth muscle cell (VSMC) contraction; and the cellular environment of the neurovascular unit. 10 Although absolute cerebrovascular tone is challenging to quantify in man, impaired responsiveness of cerebral blood flow (CBF) to stimuli has been used as a surrogate of cerebrovascular function and the interaction between endothelial cells and VSMCs. ΔCBF is commonly measured either as cerebrovascular reactivity to carbon dioxide (CVR) 11 or in response to neuronal activity (“neurovascular coupling, NVC”12,13). Autoregulation represents cerebrovascular responsiveness to blood pressure (BP) changes, targeting stable CBF as the principal mechanism protecting the brain against hypo- or hyperperfusion. 14 However, there has been a limited assessment of its role in cSVD. 15 Furthermore, although CVR, NVC, and autoregulation share a common effector pathway through VSMC contraction, the overlap between the underlying mechanisms driving VSMC contraction is less clear.
Limited research into cerebral autoregulation (CA) in cSVD reflects our limited definition of its clinical role. Despite active cerebrovascular compensation for BP fluctuations being hypothesized in the early 20th century 16 (vs. passive dependence on systemic BP) 17 and excellent empirical evidence, 18 it has little impact on clinical care, is not routinely measured, and no interventions specifically target autoregulation in cerebrovascular disease. This limited progress reflects the heterogeneity of methods measuring autoregulation and practical challenges in measurement in vulnerable patients. However, advances in the past decade are creating a paradigm shift in our understanding.
This review aims to summarize our understanding of how mechanisms underlying CA may be affected in cSVD, evidence for autoregulatory dysfunction in clinical studies of individuals with cSVD, and the potential for current autoregulation therapeutic targets and future research to improve cSVD outcomes.
Autoregulation: a matter of time
Autoregulation is the homeostatic process whereby the body maintains a stable CBF in the face of variations in BP to limit the effects of hypoperfusion and hyperperfusion. 18 Lassen’s identification of the autoregulatory plateau (Figure 1), whereby CBF remains relatively constant across a range of BP, combined reports from different patient groups utilizing Xenon-133, PET, or SPECT to derive average CBF over minutes to hours at different BPs. 19 This is now referred to as static autoregulation (sCA) and still dominates our understanding: excess hypotension directly causes hypoperfusion; stable CBF occurs across a central plateau; excess hypertension induces forced vasodilatation with a linear relationship between BP and CBF. This response to sustained BP was confirmed in sedated mammals (rats, 20 dogs, 14 monkeys 21 ), but in day-to-day life, BP fluctuations occur beat-to-beat and minute-to-minute.22–24
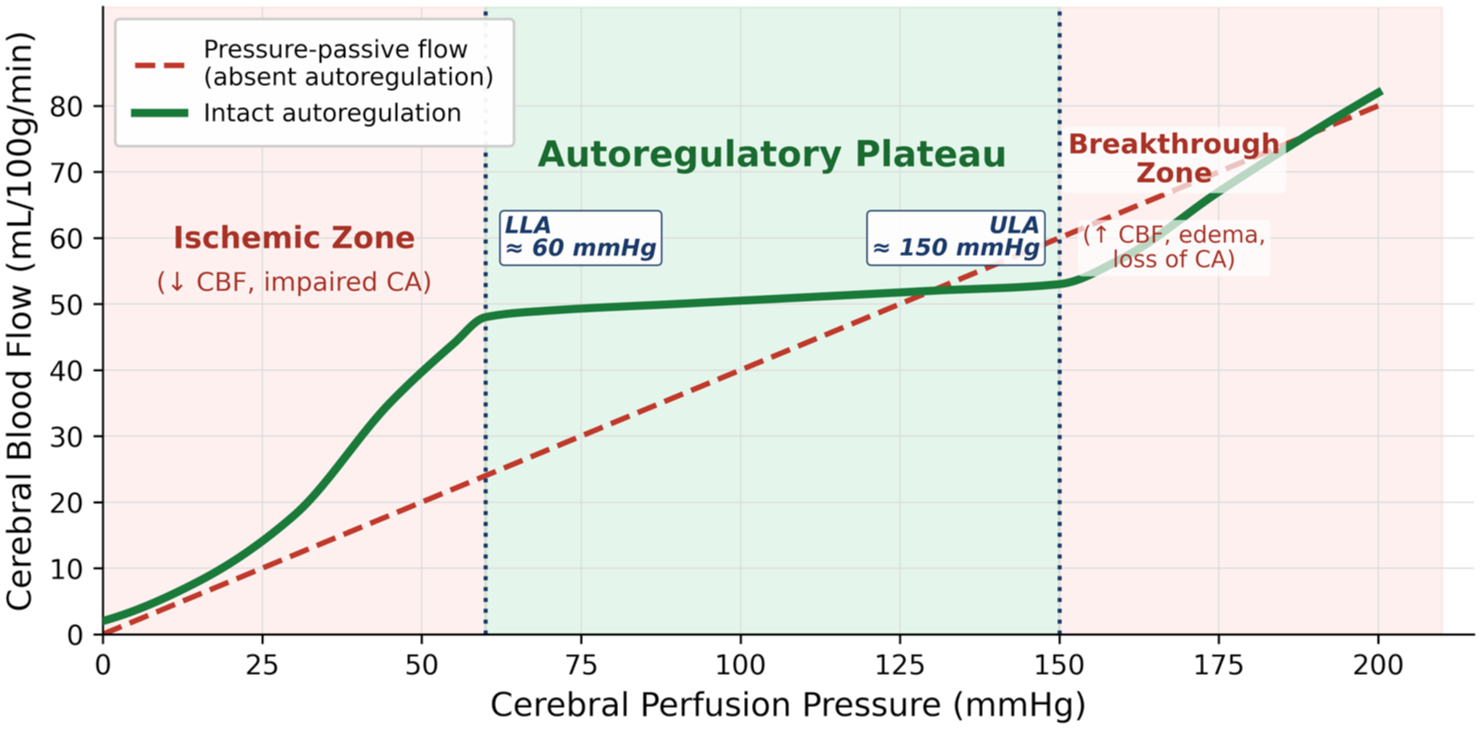
Autoregulation curve of static cerebral autoregulation. The green line represents autoregulation versus pressure-passive flow. CBF = cerebral blood flow; CA = cerebral autoregulation; LLA = lower limit of autoregulation; ULA = upper limit of autoregulation.
The advent of transcranial Doppler ultrasound (TCD) in the 1980s 25 with non-invasive BP measurement through the volume-clamp method 26 enabled measurement of beat-to-beat cerebral blood flow velocity (CBV) and BP and thus definition of dynamic CA (dCA) 14 as the magnitude and speed of CBF recovery after a change in BP (Figure 2).
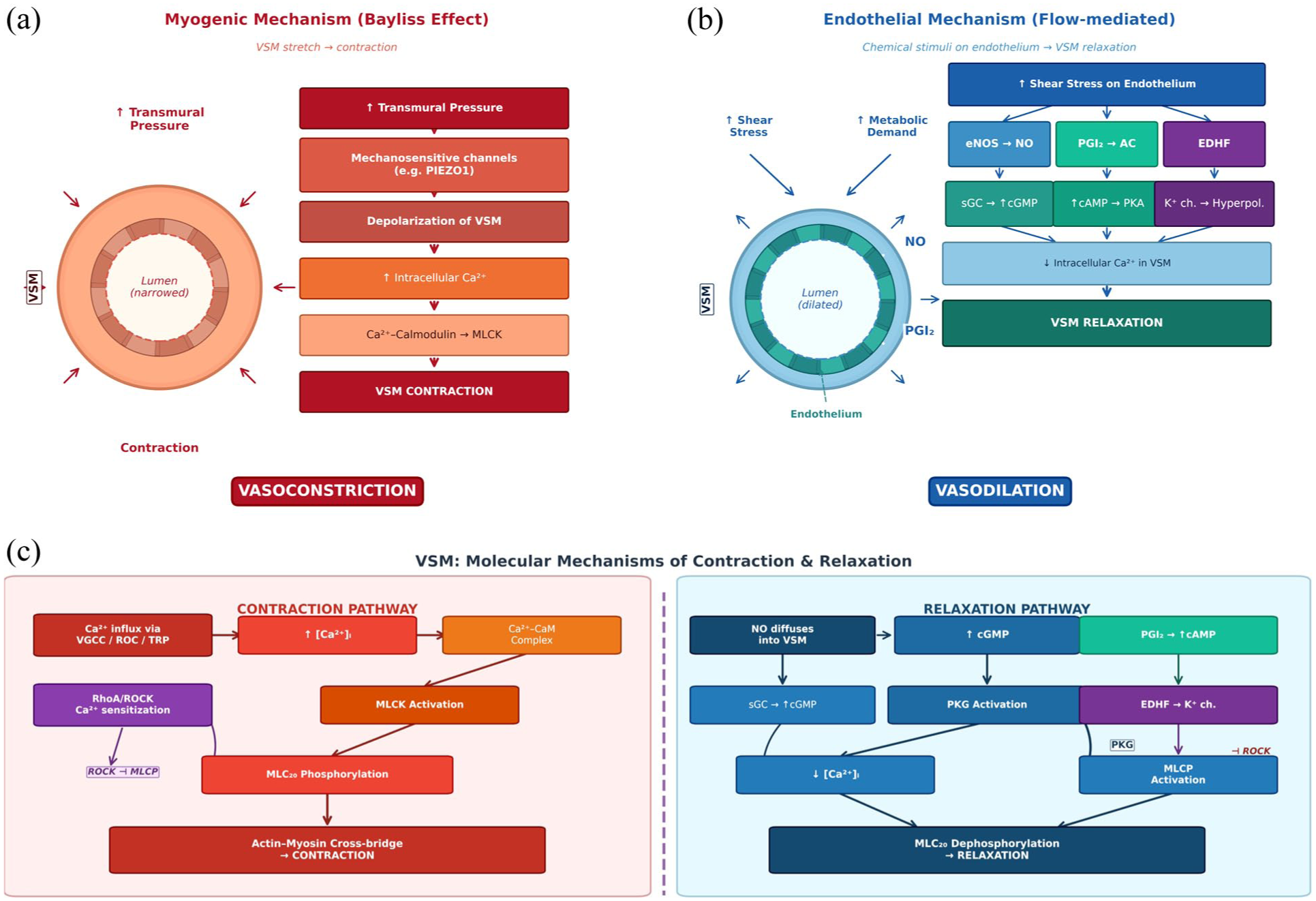
Cellular mechanisms of autoregulation at the vessel wall. (a) Myogenic mechanism (Bayliss effect): increased transmural pressure activates mechanosensitive channels (e.g. PIEZO1), leading to depolarization and calcium-dependent vasoconstriction. (b) Flow-mediated endothelial mechanism where shear stress activates signaling via the three presented pathways. (c) Molecular pathways of VSM contraction or relaxation. NO = nitric oxide; EC = endothelial cell; MLCK = myosin-light chain kinase; SMC = smooth muscle cell; sGC = soluble guanylate cyclase; MLCP = myosin-light chain phosphatase.
Each modality has its limitations. TCD cannot obtain adequate “bone windows” to measure CBV in all people, particularly older women who are at an increased risk of cSVD, and is restricted to the large vessels of the circle of Willis. This may introduce bias in the epidemiology of cSVD. Near-infrared spectroscopy (NIRS) utilizes differential infrared light absorption properties of oxygenated versus deoxygenated blood, 27 with a similar temporal resolution to TCD, but has limited tissue penetration to tissues most at-risk in cSVD. MRI has had limited temporal resolution, although new parallel imaging techniques 28 enable acquisition times at sub-second temporal resolution. This could allow estimation of dCA deep within the brain, but is yet to be applied in large populations with cSVD.
Historically, there was limited consensus on the measurement of dCA. Pharmacological challenges (phenylephrine or nitric oxide) inducing BP changes 29 could pose risks in vulnerable cSVD patients, and results may be confounded by direct effects on the cerebral circulation. 30 Mechanical methods of promoting BP changes (thigh cuff release, repeated sit-to-stand maneuvers) provide a reliable stimulus and reasonable signal:noise ratio, but their tolerance is limited in cSVD and impractical to apply in large cohorts. 31 The alternative is to use spontaneous fluctuations in BP, either spontaneously occurring or due to rhythmic oscillations. 14 This “resting state” approach produces small effect sizes and a low signal:noise relationship, and the fluctuations in BP may not be “spontaneous” but due to rhythmic oscillations in autonomic nervous system activity. 32 Nonetheless, its ready applicability has made it the most commonly used clinical technique, leading to standardized analysis approaches. 31
Quantifying autoregulation
Quantification of dCA in cSVD falls into time-domain 31 or frequency-domain methods (Figure 3). 33 Time-domain analyses relate changes in BP to changes in CBF over time, with the most widely employed index in cSVD being the correlation coefficient between ΔBP and ΔCBv (“Mxa”), which is simple to implement, and enables measurement in 3- to 5-minrolling windows, providing continuous monitoring applicable in neuro-intensive care. However, correlation oversimplifies dCA and is sensitive to physiological and measurement noise; for example, a low Mxa may reflect either efficient autoregulation or poor signal quality. 34 Finally, cerebrovascular responses to a change in BP are not instantaneous, yet Mxa is derived without correcting for any time-lag.
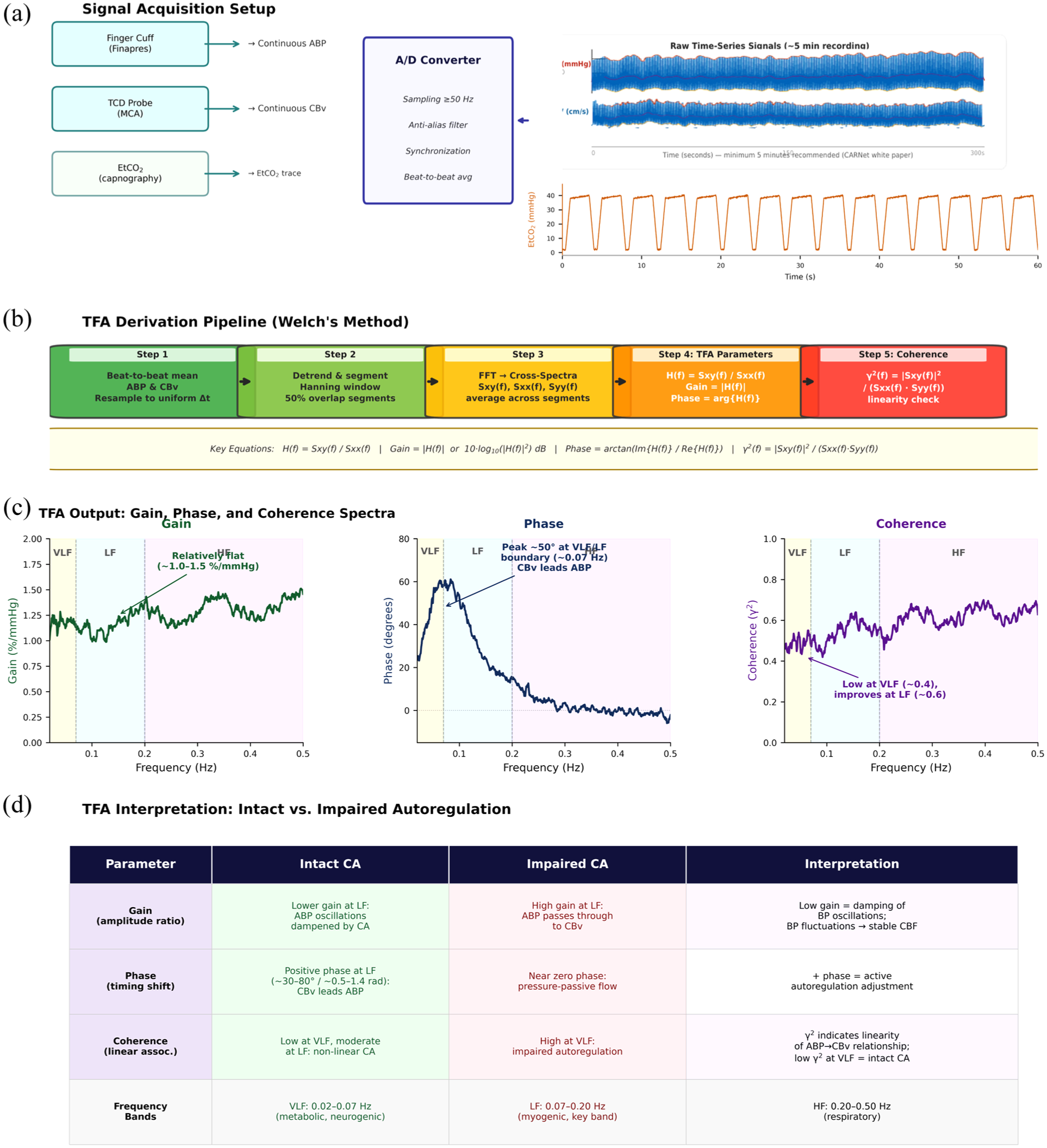
Calculating autoregulation by transfer function analysis (TFA). (a) Data acquisition; (b) Primary analysis; (c) Example results of transfer function analysis (d) Interpretation. Validity is checked by the 95% confidence limit of coherence. A/D = analog-digital; MCA = middle cerebral artery; ABP = arterial blood pressure; VLF = very low frequency; LF = low frequency; HF = high frequency; etCO2 = end-tidal CO2; Sxx(f) = autospectral density.
The autoregulatory index (ARI) models the CBF response to a step change in BP, using a second-order difference equation, matching the observed response to 10 predefined curves that vary from 9 (rapid return of CBF) to 0 (autoregulation is absent). ARI was originally developed using induced BP changes, but can be estimated from resting beat-to-beat monitoring by modeling impulse responses in a linear input–output framework. 31 However, fitting the resulting response function can be problematic, requiring manual review to exclude unphysiological results, 34 and model instability. 34 Finally, the speed of return of CBF after a step response is captured by indices such as rate of recovery. More sophisticated techniques, largely available in specialist centers, capture more complex features such as multimodal pressure flow (MMPF); 14 while analysis differentiating responses to BP increases and decreases can clarify differential responsiveness of vasoconstrictor and vasodilator mechanisms, and thus myogenic and endothelial responses. 35
Frequency-domain analyses commonly use transfer function analysis (TFA), using Fourier’s theorem to represent the relationship between BP and CBF as a sum of sine waves at different amplitudes and phase delays across frequency bands. Gain reflects the amplitude of transmission, while phase reflects temporal delay and coherence, the strength of the relationship (comparable to R2), with coherence often used to define when gain and phase are reliably estimated. 14 TFA has limitations, particularly in older patients at greater risk of cSVD in whom recording can be challenging, resulting in low signal quality and coherence, as well as violation of the assumption of “stationarity” (a stable mean, variance, and autocorrelation over time). Finally, TFA models dCA as a linear time-invariant model relating BP as an input function and CBv as an output function. However, in the presence of confounding mechanisms that synchronously affect both BP and CBF (such as PaCO2), this assumption is violated. More complex methods of analysis are under development to overcome these limitations, but may be less accessible in practice.
Mechanistic overlap between autoregulation and cSVD
The mechanisms driving autoregulation are directly implicated in cSVD. First, autoregulation acts on the BP waveform transmitted from the aorta, through large extracranial vessels and the circle of Willis to reach the perforators, cortically destined pial vessels, arterioles, and capillaries where autoregulation takes place. 8 cSVD is strongly associated with abnormalities of this input signal (BP), including hypertension,36,37 aortic stiffness,38–40 cerebral arterial pulsatility, 40 impaired baroreceptor sensitivity,24,41 and BP variability. 42 These factors result in greater amplitude and variability of transmitted BP, presenting a greater challenge to autoregulatory mechanisms and directly influencing dCA analysis methods. 24
Second, cSVD pathophysiology affects the vasculature that mediates autoregulation. CBF is principally regulated by cerebrovascular tone, with flow proportional to the fourth power of vessel radius (Poiseuille’s law), and thus, small changes in arteriolar caliber have large effects on resistance. Arteriolar VSMCs and contractile, capillary pericytes are the principal sites of myogenic tone and autoregulation.10,43 VSMC vasodilatory function can be tested using CVR to carbon dioxide, which is significantly reduced in cSVD, 11 independent of changes in systemic arterial stiffness, 11 representing a key therapeutic target.44–47 CVR is mediated by reduced pH within and outside VSMCs and thus may not be affected by dysfunction of the sensor components of autoregulation, but would still be predicted to affect the effector mechanism. 48 This is also suggested by impaired vasodilatation to neuronal activity, 12 associated with cSVD-dependent cognitive dysfunction. 49 However, there is evidence of independence of dCA and CVR in cSVD. 50
The principal mechanism of autoregulation is the myogenic response, whereby intravascular pressure induces depolarization of VSMCs via mechanotransducers on the cell membrane, which induce calcium entry and activate the RhoA-ROCK pathway, resulting in phosphorylation of myosin and vasoconstriction. This reflex normalizes arterial wall tension, compensating for short-term changes in cerebral perfusion pressure, and thus driving the sCA plateau and the dCA response. It occurs in isolated vessel preparations and vessels denuded of endothelium, consistent with a pure VSMC-dependent function. 51 Direct evidence of impaired mechanotransduction in cSVD is limited. However, myogenic tone is abnormal in monogenic models of CADASIL, 52 and in models of sporadic cSVD, including the spontaneously hypertensive rat, 53 angiotensin-II, 54 and chronic cerebral hypoperfusion 55 models.
The myogenic response occurs within a complex regulatory environment that modifies resting cerebrovascular tone and responses to stimuli. Most powerfully, dCA responses are modified during hypercapnia.56,57 Second, endothelial release of nitric oxide is the dominant chemical determinant of vascular tone, diffusing into VSMCs to induce vasodilatation. Its precise role in directly mediating autoregulation over longer timescales than the myogenic response is uncertain. However, abnormalities of NO metabolism can shift the autoregulatory curve to the right, 58 while loss-of-function mutations in NO-synthase prompt an impaired cerebral autoregulatory response. 59 The role of antagonistic vasoconstrictors (angiotensin, endothelin-1) has not been tested.
There is extensive innervation of the large cerebral vessels by sympathetic fibers via the stellate ganglion and sympathetic chain. Although extrinsic innervation is sparse in the deep perforators affected in cSVD, intrinsic autonomic innervation persists within the brain. Sympathetic blockade results in excessive CBF responses to elevated BP and impaired autoregulation measured by TFA. 60 However, autonomic dysfunction increases BP variability, 24 affecting estimates of dCA, indicative of the challenge in separating systemic BP from cerebrovascular effects. However, given the strong association between cSVD and impaired autonomic function, these interactions likely influence dCA and may represent therapeutic targets.
Finally, major cardiovascular risk factors36,61 directly overlap with impaired autoregulation. First, age is the strongest determinant of sporadic cSVD. dCA phase is often preserved with age, but autoregulatory gain is reduced, 62 although this may reflect lower CBv rather than myogenic failure. Second, long-standing hypertension is the strongest risk factor for cSVD, 36 reflecting the transition from mid- to late-life hypertensive phenotypes. 63 While autoregulation is an intrinsic response to acute hypertension or hypotension, chronic hypertension induces a shift in the static autoregulatory curve to the right in animal studies and clinical populations. 64 Finally, diabetes is associated with both cSVD and impaired autoregulation. 65
Autoregulation in sporadic cSVD
There are limited cross-sectional population-based studies of autoregulation in cSVD, with no sufficiently powered longitudinal studies. The largest identifiable study in 113 neurology outpatients with incidental cSVD on MRI reported increased TFA-gain (at both 0.02–0.07 and 0.07–0.2 Hz) compared with 83 controls, 66 including after adjustment for age and sex. There was furthermore a correlation between phase (LF) and the total cSVD score, total WMH, deep and periventricular WMH individually, lobar microbleeds, and severe ePVS. However, the independent association with each imaging marker was unclear, and the association with lobar microbleeds may indicate overlap in the underlying pathology (e.g. CAA vs. sporadic cSVD). Nonetheless, a similar reduction in phase was reported in smaller studies with increasing DTI indices of microstructural injury in 48 largely white participants with vascular risk factors, 67 and in 14 individuals with WMH, although again this was potentially mediated by amyloid pathology. 68 Finally, reduced phase modulated the association between deep medullary veins and WMH in 95 people with imaging markers of cSVD. 69 Further evidence comes from a large retrospective cohort of 346 patients undergoing cardiopulmonary bypass with continuous TCD-based autoregulation monitoring. Impaired autoregulation (Mxa ⩾ 0.4 occurred in 32% of patients and was independently associated with MRI-defined small vessel disease (WMH), but not with age or moderate to severe large vessel stenosis. These findings support a specific link between cSVD pathology and impaired pressure-flow regulation, independent of proximal arterial disease.
Autoregulation in patients with cSVD after stroke
After a lacunar stroke, 71 participants in a Chinese cohort showed reduced phase (within 0.06–0.12 Hz) in both ipsilateral and contralateral hemispheres, and in posterior cerebral arteries, compared with controls, 70 consistent with smaller studies. 71 Furthermore, a number of studies reported either decreased phase72–76 or impaired autoregulation by ARI,75–78 associated with more severe WMH after territorial ischemic strokes due to either embolic 75 or mixed aetiologies.50,72,77 This usually reflected global impairment of autoregulation, 50 present even in mild strokes78,79 implying an underlying functional deficit rather than a sequelae of the stroke. Although some studies identified greater impairments in the territory 75 or ipsilateral to larger infarcts, there were more global deficits in lacunar stroke. 71 However, most commonly, there were limited differences between stroke subtypes, with associations more closely related to WMH burden or stroke severity. 77
The global nature of dCA impairment in stroke patients with cSVD may reflect either established vasculopathy or impairments of systemic BP control. For example, impaired dCA has been associated with severe hypertension, 80 renal impairment, 73 and sympathovagal dysfunction in the acute phase of stroke. 81 This makes attribution of impaired autoregulation solely to cSVD pathology difficult. In contrast, impaired dCA is also associated with a higher probability of complications of stroke, including hemorrhagic transformation and oedema. 82 Whether this relationship is mediated by coexistent cSVD, acute infarct characteristics, or systemic hemodynamics instability is uncertain.
Autoregulation in specific forms of cSVD
In sporadic disease, disentangling the effect on dCA of established vasculopathy versus risk factors is difficult. However, in monogenic CADASIL rodent models without hypertension, autoregulation was impaired, 52 although there was no evidence of autoregulatory dysfunction in 25 patients with CADASIL 83 or with WMH severity in 10 individuals with Fabry’s disease. 84 One abstract reported “autoregulatory” dysfunction in nine individuals with CADASIL, 85 but this reflected the autonomic response to isometric handgrip. This is in contrast to reduced cerebrovascular reactivity in the Fabry’s group 84 and in CADASIL patients using either TCD 86 or within WMH on MRI. 87 This may reflect that the vasodilatory dysfunction in small vessels is consistently demonstratable with CVR compared with large artery-dependent autoregulation.
Cerebral amyloid angiopathy (CAA) is a sporadic cSVD defined by a primary small vessel vasculopathy without associated cardiovascular risk factors. However, there are few studies. One study reported that, in 29 patients, more cerebral microbleeds or superficial siderosis were related to reduced phase in the posterior cerebral artery. 88 This evidence gap reflects that most studies in CAA populations focus on MRI imaging of the brain, with little rationale to assess large cerebral vascular function.
Autoregulation in vascular cognitive impairment
Most autoregulation studies in cSVD have focused on imaging markers of cSVD, with limited studies assessing effects on the ischemic sequelae of cSVD or cognitive function. In contrast, due to its higher prevalence, more studies have been in of Alzheimer’s disease (AD). Although AD is associated with impaired cerebrovascular function, 89 including reduced cerebrovascular reactivity and impaired neurovascular coupling, the evidence for impaired autoregulation in AD is sparse. Limited studies identified impaired autoregulation,90,91 or used composite indices of cerebrovascular function dominated by CVR. 90 Overall, a recent meta-analysis demonstrated no association between autoregulatory function and AD or mild cognitive impairment, 92 with a single study even suggesting worse dCA efficiency in healthy individuals than in mild cognitive impairment, 93 also consistent with a study in mild cognitive impairment (MCI). 94 Unfortunately, studies addressing MCI did not discriminate between neurodegenerative and vascular causes. Similarly, very limited reports suggest reduced phase (VLF) may be associated with post-stroke cognitive decline, but lobar microbleeds predicted cognitive decline, implying an amyloid-dependent mechanism 95
Hemodynamic interventions in cSVD
The theoretical and empirical relationship between autoregulatory dysfunction and cSVD supports the potential of autoregulation as a therapeutic target. However, no current medicinal interventions modify dCA in isolation from other hemodynamic measures. Direct targeting of autonomic function and tone (e.g. baroreceptor stimulator or vagal nerve stimulation) is feasible but is hard to justify in cSVD without better observational evidence. Nonetheless, current hemodynamic interventions for cSVD are potentially informative. First, in SPRINT-MIND, intensive BP control (<120/70) was associated with reduced cognitive impairment 96 and a reduction in progression of WMH, 97 although autoregulation was not directly measured. In contrast, in TREAT-SVDs, there was no effect of the commonly used antihypertensives on cerebrovascular vasodilatory function, except in a limited number of participants with CADASIL. 98
Nonetheless, vasodilators have shown promise. In 56 LACI-1 patients with a previous subcortical infarct, treatment with vasodilators (cilostazol or ISMN) was associated with improved vasodilatory function, 30 while LACI-2 demonstrated in 363 participants that 1 year of isosorbide mononitrate (ISMN) was associated with reduced cognitive decline. 47 Although not testing autoregulation, these medications did target improved cerebrovascular tone with an apparent association with disease outcomes.
Finally, PDE5 inhibitors, which reduce breakdown of cGMP in VSMCs following NO release, also show promise in cSVD. The OxHARP trial11,45,99 demonstrated that CVR and cerebral pulsatility were independently associated with cSVD, 11 while treatment with sildenafil improved CVR and absolute CBF on TCD and MRI. Similarly, the ETLAS 1 trial 100 demonstrated improvements in cerebral oxygenated hemoglobin levels on NIRS with tadalafil, while the phase-2 ETLAS-2 trial 101 demonstrated an improvement in cortical CBF, if not CVR.
Overall, there are drugs that enhance VSMC function via vasodilatory pathways that are currently the most promising treatments in cSVD. Whether any clinical benefits are mediated by effects on common pathways that underlie autoregulation, or due to enhanced vasodilatory reserve, or a combination of mechanisms, remains to be determined.
Future research requirements
Further research is essential before autoregulation can be positioned as a therapeutic target in cSVD. First, more studies measuring autoregulation in cSVD are essential to differentiate which cSVD imaging markers are associated with impaired autoregulation, and whether it is primarily a sequelae of the disease or mediates the effect of risk factors on the brain. Second, methodological consensus is essential. Currently, a wide range of methods are employed despite attempts to standardize the approach. Third, longitudinal studies are crucial to understand whether autoregulatory dysfunction predicts the progression of vascular injury. Fourth, methods of measurement need improvement. Currently, virtually all studies use TCD that assess large proximal arteries, but the pathology of the disease affects distal perforators and capillaries. Therefore, more subtle dCA dysfunction may not be identifiable with TCD but will require high temporal resolution MRI or multimodal physiological modeling.
Conclusion
CA is critical to the control of blood flow to the brain. It is underlying mechanisms overlap with key sites of pathophysiology in small vessel disease, and autoregulatory dysfunction is associated with disease severity. However, clinical studies are very limited, and any causative role for autoregulation is unclear. Although promising vasodilator drugs for cSVD target some of these overlapping pathways, their effects on autoregulation are unknown. Therefore, although autoregulation has strong potential as a therapeutic target in cSVD, extensive further research is required to understand this potential.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: AJSW has previously received consultancy fees from Woolsey Pharmaceuticals and has current research collaborations with Medtronic and Aribio Ltd.
Funding
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article: AW is funded by an MRC-NHMB Project Grant (MR/Y014634/1), the St. Mary’s Development Trust, and a Stroke Association Senior Clinical Lectureship (SCLfMP25\100006). OLL is funded by a Stroke Association fellowship (SAPDF 21\100029).