
Abstract
Background:
High-quality randomized controlled trials (RCTs) have extended the endovascular therapy (EVT) time window to 24 h post-stroke onset in selected patients with acute ischemic stroke (AIS). Recent retrospective studies indicate that EVT performed beyond 24 h may still improve clinical outcomes. However, the specific benefit–risk profile in this ultra-late window remains unclear.
Aim:
The trial is designed to address this evidence gap and determine the benefit–risk balance of EVT in the ultra-late window.
Methods and design:
The Large Artery occlusion Treated in Extended Time with Mechanical Thrombectomy (LATE-MT) is an investigator-initiated, multicenter, prospective, randomized, open, blinded-endpoint assessment (PROBE) clinical trial. The trial adopted an adaptive group-sequential design, recruiting 336 AIS patients with large-vessel occlusion within 24–72 h of the last known well across 35 stroke centers in China. Eligible subjects who meet both clinical and imaging selection criteria are randomized 1:1 to EVT or medical management.
Outcomes:
The primary outcome is an ordinal shift analysis of scores on the modified Rankin scale (mRS) at 90 days. Key secondary outcomes include neurological function at 24 h and 7 days, death/major disability, and utility-weighted mRS (UW-mRS) at 90 days. Safety outcomes include any intracranial hemorrhage (ICH), symptomatic ICH, serious adverse event, and all procedural complications.
Introduction and rationale
Endovascular therapy (EVT) is the primary treatment for acute ischemic stroke (AIS) with large-vessel occlusion (LVO). The DAWN and DEFUSE3 trials have extended the EVT treatment window from 6 to 24 h.1,2 However, nearly half of AIS patients arrive beyond 24 h due to prehospital delays, especially in developing countries, missing the optimal treatment window.3–5 Some with good collateral circulation may maintain salvageable penumbra beyond that period.6,7 In DEFUSE3, 18% of controls continued to meet imaging criteria at a median of 38 h but had extremely poor outcomes. 7
The benefits and harms of EVT performed beyond 24 h are unproven. With growing attention, several observational studies have been reported.8–11 A recent meta-analysis summarized 10 studies indicating that EVT was associated with improved functional outcomes, despite a higher risk of symptomatic intracranial hemorrhage (sICH). 12 A well-designed randomized controlled trial (RCT) is needed to determine the effectiveness and safety of EVT in the ultra-late window.
To address this evidence gap, we have designed the Large Artery occlusion Treated in Extended Time with Mechanical Thrombectomy (LATE-MT) trial to determine the balance of potential benefits and risks of performing EVT in selected AIS patients, including identifying a responder subgroup that stands to benefit the most.
Methods
Design
LATE-MT is an adaptive group sequential design conducted, multicenter, prospective, randomized, open, blinded-endpoint assessment (PROBE) clinical trial. The study design is outlined in Figure 1. Ethics committee approval has been obtained for each participating center.
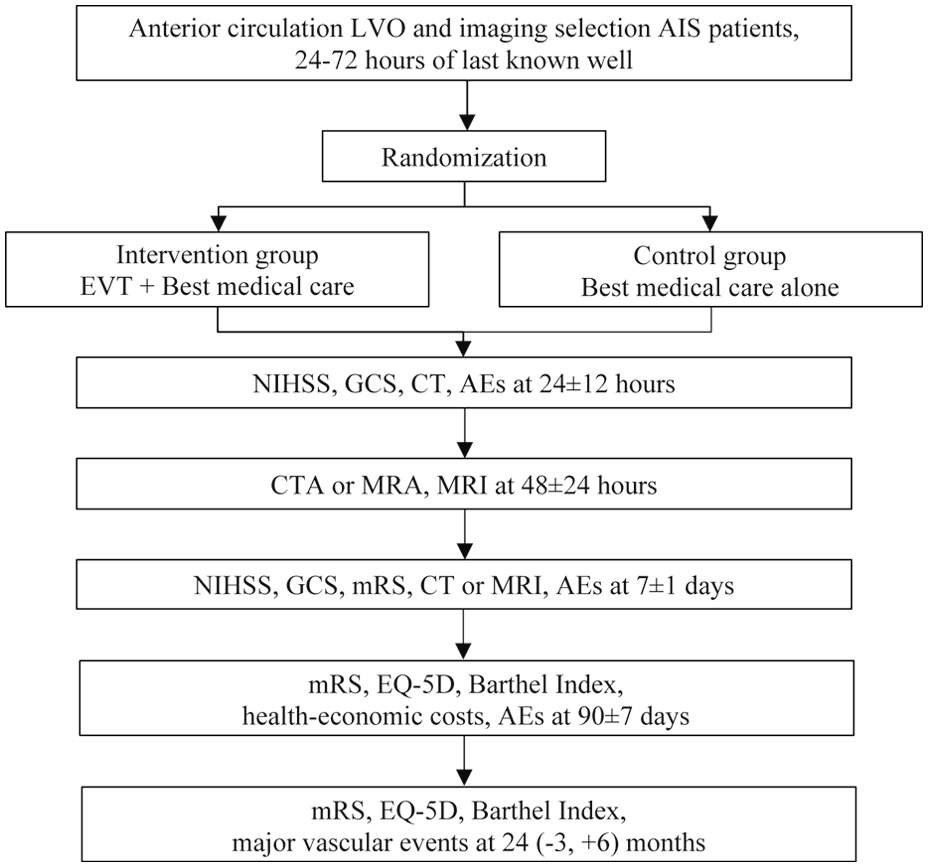
Flowchart in the LATE-MT trial.
Population
All stroke patients with suspected LVO who present to participating hospitals within 24–72 h of their last known well will be screened for eligibility. Eligible patients may be admitted directly to sites, transferred from other hospitals, or experience an in-hospital progressive stroke. It is recommended to perform a multimodal imaging examination to determine the location of vascular occlusion and perfusion parameters. The UGuard® software (Union Strong Tech, Beijing) is used to quickly obtain quantitative results for ischemic core and mismatch volume. 13 Eligible patients were those aged 18 years or older, presenting 24–72 h after stroke onset or last known well, with anterior circulation LVO from extracranial internal carotid artery to M2 segment, an National Institutes of Health stroke scale (NIHSS) score of ⩾ 6 at randomization, an ischemic core volume of < 50 mL, a mismatch ratio of ⩾ 1.8, a mismatch volume of ⩾ 15 mL, and who provided written informed consent from themselves or legal representatives. Key exclusion criteria include allergy or contraindication to EVT-related agents/procedures, EVT already attempted, ICH on imaging, previous stent in a target vessel, and occlusions in multiple vascular territories. Detailed inclusion and exclusion criteria are provided in the Supplemental Materials.
Randomization
After confirming eligibility, patients will be randomized through a central, Internet-based, password-protected system at a 1:1 ratio to the intervention group or control group. The randomization sequence was generated using a minimization algorithm that balanced time from symptom onset to randomization (dichotomized as 24–48 h vs 48–72 h) between the two treatment groups. All subjects must be within 24–72 h from symptom onset to randomization. Investigators and clinicians should not delay or accelerate the enrollment period to increase or avoid enrolling patients.
Intervention
Patients are assigned to either intervention (immediate EVT) or control (only standard care). The intervention group should undergo EVT as soon as possible, with the time from randomization to arterial puncture ideally kept under 60 min. The trial does not restrict the choice between stent or aspiration thrombectomy, or the number of procedures performed. Operators may choose direct angioplasty. Any National Medical Product Administration (NMPA)-approved devices are allowed. The control group must avoid any EVT-related procedures, including arterial puncture and diagnostic angiography alone, and receive only standard medical care according to local guidelines.
Outcomes
The primary outcome is an ordinal shift analysis of scores on the modified Rankin scale (mRS) at 90 days. Secondary outcomes include (1) neurological function, measured by NIHSS scores at 24 h and 7 days/discharge; (2) death or major disability (mRS 3–6), separately on death and disability (mRS 3–5), and utility-weighted mRS (UW-mRS) at 90 days; (3) health-related quality of life using the EuroQol Group 5-Dimension Self-Reported Questionnaire (EQ-5D) at 90 days; (4) Barthel Index at 90 days; (5) duration of hospitalization; and (6) hospital service costs. In addition, there are two imaging endpoints: follow-up infarct volume, and recanalization assessed by computed tomography angiography (CTA) or magnetic resonance angiography (MRA).
Safety outcomes include (1) any ICH within 7 days (Heidelberg Bleeding Classification); 14 (2) sICH within 48 h (Heidelberg Bleeding Classification); 14 (3) all severe adverse events during follow-up; and (4) procedural complications.
On 1 May 2025, the protocol was amended to include extended follow-up procedures. Patients who were alive at the 90-day follow-up underwent a 24-month follow-up for long-term outcome assessment, including scores on the mRS, EQ-5D questionnaire, and Barthel Index, as well as the occurrence of major vascular events.
Data Safety Monitoring Body
An independent, expert Data Safety Monitoring Body (DSMB) will monitor efficacy and safety for early dramatic benefits and harmful effects. The DSMB will be governed by a charter that outlines their responsibilities, procedures, and confidentiality. The first meeting will be held before study enrollment begins. During the trial, the DSMB will meet at prespecified time points to review unblinded data and check drop-out rates, event rates, and outcome differences between groups. It will provide the Trial Steering Committee (TSC) with reports and recommendations to continue, pause, or terminate recruitment after each meeting. The TSC will then fully review these recommendations and render the final decision.
Sample size calculation
The study is designed to have 90% power to detect a 2.0 odds ratio (OR) for improved functional recovery at 90 days between groups. Ordinal logistic regression will be used for analysis. Based on DEFUSE3, the control group mRS distribution is assumed to be 8.0%, 4.0%, 4.0%, 16.0%, 27.0%, 16.0%, and 26.0% (rounded) for scores 0–6, respectively. 2 A sample size of 276 subjects is estimated to show this effect with 90% power and 5% type 1 error.
To account for intervention effect uncertainty, we will use an adaptive group-sequential design. We assume 2-sided Lan-DeMets error spending functions of the O’Brien-Fleming type. 15 This increases the sample size to 294. With two interim analyses, simulations show a 69% chance of stopping for futility at the second interim if OR = 1 (null). If OR = 2 (alternative), the chance of stopping for efficacy is 5% at the first interim and 55% at the second interim. We assume up to 3% missing data and 5% crossovers. Thus, the target sample size is 336. An unblinded sample-size re-estimation will occur at the second interim, using the promising zone approach by Mehta and Pocock. If the conditional power is between 33% and 80%, the sample size will increase to reach 80% conditional power. The minimum sample size is 336, and the maximum is 672. If conditional power is below 33% or above 80%, the sample size remains 336, unless efficacy or futility boundaries are crossed.
Statistical analysis
The intention-to-treat (ITT) principle will be applied to the primary analyses. Baseline characteristics will be summarized by treatment group. The primary endpoint will be analyzed using logistic regression adjusted for stratification variables. Sensitivity analyses will include additional covariate adjustments and different assumptions about missing data to assess robustness. For the secondary endpoints, the family-wise error rate will be controlled using the Holm-Šidák correction. A separate statistical analysis plan (SAP) will be developed for the 24-month long-term outcomes.
Heterogeneity of treatment effect on the primary endpoint will be assessed across subgroups by adding an interaction term to the model. The prespecified subgroups include age, sex, time from onset to randomization, determination of time of stroke, baseline NIHSS, occlusion location, ischemic core volume, and stroke etiology. Details of all analyses were specified a priori in a full SAP (available online at https://osf.io/bsgu4/files/t9fxv).
Discussion
LATE-MT is the first RCT to investigate the efficacy and safety of EVT in AIS patients beyond the 24-h window. The results of this study will provide evidence regarding whether EVT benefits for patients with LVO who experience prehospital delays or progressive stroke.
The trial follows the approach of the DAWN and DEFUSE3 studies, using perfusion imaging to select patients with small ischemic core volume and salvageable penumbra.1,2 This selection criterion was pivotal to the positive outcomes of both studies. Before study initiation, despite limited data, we observed that ultra-late window EVT meeting DAWN inclusion criteria yielded favorable outcomes comparable to those of the DAWN trial group. 16 A Korean study indicated that only ultra-late window patients meeting DEFUSE3 and DAWN criteria derived benefit from EVT or showed a more pronounced trend. 8 Given the considerable uncertainties and the potentially higher risks associated with the ultra-late time window, this study applied stricter ischemic core volume requirements than DEFUSE3. As the RAPID software has not obtained a license in China, it is required that a commonly used, NMPA-approved UGUARD software be uniformly adopted to avoid heterogeneity. This software has shown strong volumetric agreement with RAPID software in a validation study. 13
The trial set the upper limit of the time window at 72 h post-stroke onset. This is because very few patients experience such prolonged prehospital delays. In addition, we assumed inclusion of stroke patients experiencing early neurological deterioration (END) beyond 24 h post-onset, with three-quarters of END occurring within 72 h. 17 Setting an overly broad time window would result in sparse enrolment of very late-stage cases, leading to conclusions lacking sufficient statistical power.
The EVT strategy was not subject to excessive restrictions. Differing from previous studies, direct balloon dilatation or stenting was permitted. While stroke etiology within the 24-h window is typically cardioembolic, intracranial atherosclerosis (ICAS) accounts for a very high proportion in the ultra-late window, particularly in Asian populations.18,19 Evidence for EVT strategies for ICAS-LVO remains limited. Several studies suggest that direct balloon dilatation or stenting achieves high reperfusion rates and superior functional outcomes in ICAS-LVO lesions with minimal or no thrombus.20,21
Finally, safety events, particularly sICH, remain our special focus. Although prior studies have not demonstrated increased safety issues, the overall sample size remains limited. Recent research indicates that while EVT in the ultra-late window offers clear benefits, it is associated with a higher risk of sICH. Consequently, high-frequency imaging examinations and safety monitoring will be implemented.
Supplemental Material
sj-docx-1-wso-10.1177_17474930261458848 – Supplemental material for Large Artery occlusion Treated in Extended Time with Mechanical Thrombectomy (LATE-MT): Protocol for a multicenter randomized clinical trial
Supplemental material, sj-docx-1-wso-10.1177_17474930261458848 for Large Artery occlusion Treated in Extended Time with Mechanical Thrombectomy (LATE-MT): Protocol for a multicenter randomized clinical trial by Hongjian Shen, Pengfei Yang, Lili Song, Yongwei Zhang, Xiaoxi Zhang, Yang Zhao, Lijun Wang, Pengfei Xing, Lei Zhang, Craig S Anderson and Jianmin Liu in International Journal of Stroke
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Health Commission Capacity Building and Continuing Education Center 2021 Neurosurgical Program (GWJJ2021100204), Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0539900), Shanghai Municipal Health Commission Clinical Research Special Project (20224Z0008), Clinical Medical Research Special Project of the First Affiliated Hospital of Naval Military Medical University (2024LYA01), and Union Strong Tech (Beijing) Co., Ltd.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.