
Abstract
The pathogenesis of ulnar polydactyly in humans is not known. There are numerous syndromes that are associated with ulnar polydactyly. We have noted that the genetic defects in these syndromes lead to a disturbance of the normal balance between the two forms of the Gli3 protein (the active and repressor forms of Gli3, which are known as Gli3-A and Gli3-R, respectively), leading to a relative increase in the Gli3-R protein. We offer the hypothesis of a unified pathogenesis of ulnar polydactyly through the relative predominance of Gli3-R.
Introduction
Ulnar polydactyly (UP) is an error of the antero-posterior axis of limb development. The sonic hedgehog homolog (Shh) protein is the main controller of this axis. Deficiency of Shh leads to ulnar ray deficiency, while increased activity or over-expression of Shh leads to radial polydactyly. Although previous authors (Oberg et al., 2010) have attributed UP to defects related to failure of ‘hand-plate’ axis differentiation, the actual pathogenesis of UP in humans is not known.
One way to understand the pathogenesis of UP in humans is to investigate the effects of known gene mutations of human syndromes in which UP is a prominent feature. There are numerous syndromes that are known to be associated with UP (Goetz and Anderson, 2010). We have noted that the genetic defects in these syndromes lead to a disturbance of the normal balance between the two forms of the Gli3 protein (the active and repressor forms of Gli3, which are known as Gli3-A and Gli3-R, respectively), leading to a relative increase in the Gli3-R protein.
We offer the hypothesis of a unified pathogenesis of UP through the relative predominance of Gli3-R. We explain our hypothesis by giving a brief description of the processing of Gli3 protein and then by demonstrating how the genetic defects of syndromal UP lead to a predominance of the repressor form of the Gli3 protein.
Processing of the Gli3 protein
Shh has a key role in the antero-posterior axis of limb development. It is localized in the posterior mesenchyme of the limb bud in an area known as the zone of polarizing activity. Shh mediates the processing of Gli3 into the active form. Therefore, the normal autopod (the future hand or foot) of the limb bud will have a balanced gradient between the two forms of the Gli3 protein. Gli3-A predominates posteriorly (where Shh is located) and Gli3-R predominates anteriorly (where there is no Shh activity) (Figure 1). The processing of Gli3 by the Shh ligand occurs at the primary cilium, a slim microtubule-based organelle that projects from the surface of embryonic cells (Cardenas-Rodriguez and Badano, 2009) (Figure 2). It is composed of three parts: basal body proteins (a base that is attached to the apical actin network of the cell); the axoneme (a projecting part that is made of nine microtubule doublets with or without a pair of central microtubules); and the ciliary membrane (the cell membrane around the axoneme). ‘Cargo’ proteins, such as Gli3, are processed in the primary cilium via intraflagellar protein transport (IFT). First, the protein is transported from the base to the tip (antegrade IFT) and this is mediated by the kinesin-2 motor and IFT A/B protein complexes. The dynein motor is also attached to the IFT proteins, and is required for retrograde IFT. ADP-ribosylation factor-like 13B (ARL13B), localizes to the cilia and is required for the normal axoneme structure. Transmembrane protein 216 (TMEM 216) also localizes to the cilium and is required for the normal function of cilia (Lee et al., 2012) (Figure 2). It should also be noted that the dynein motor is made up of dynein 2 heavy chain 1 (DYNC2H1) and dynein 2 light intermediate chain 1 (DYNC2LI1). Knowledge of these structural proteins of the cilia is important because their corresponding genes are mutated in syndromes with UP, as will be discussed later.
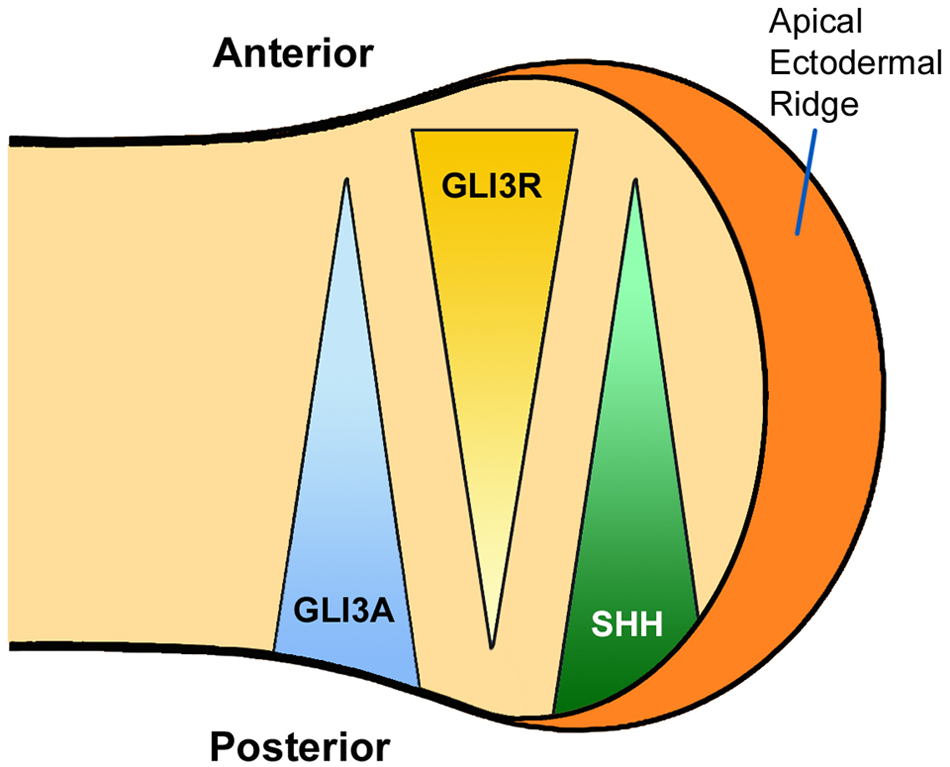
The balanced gradients of Gli3-A and Gli3-R in early limb bud development.
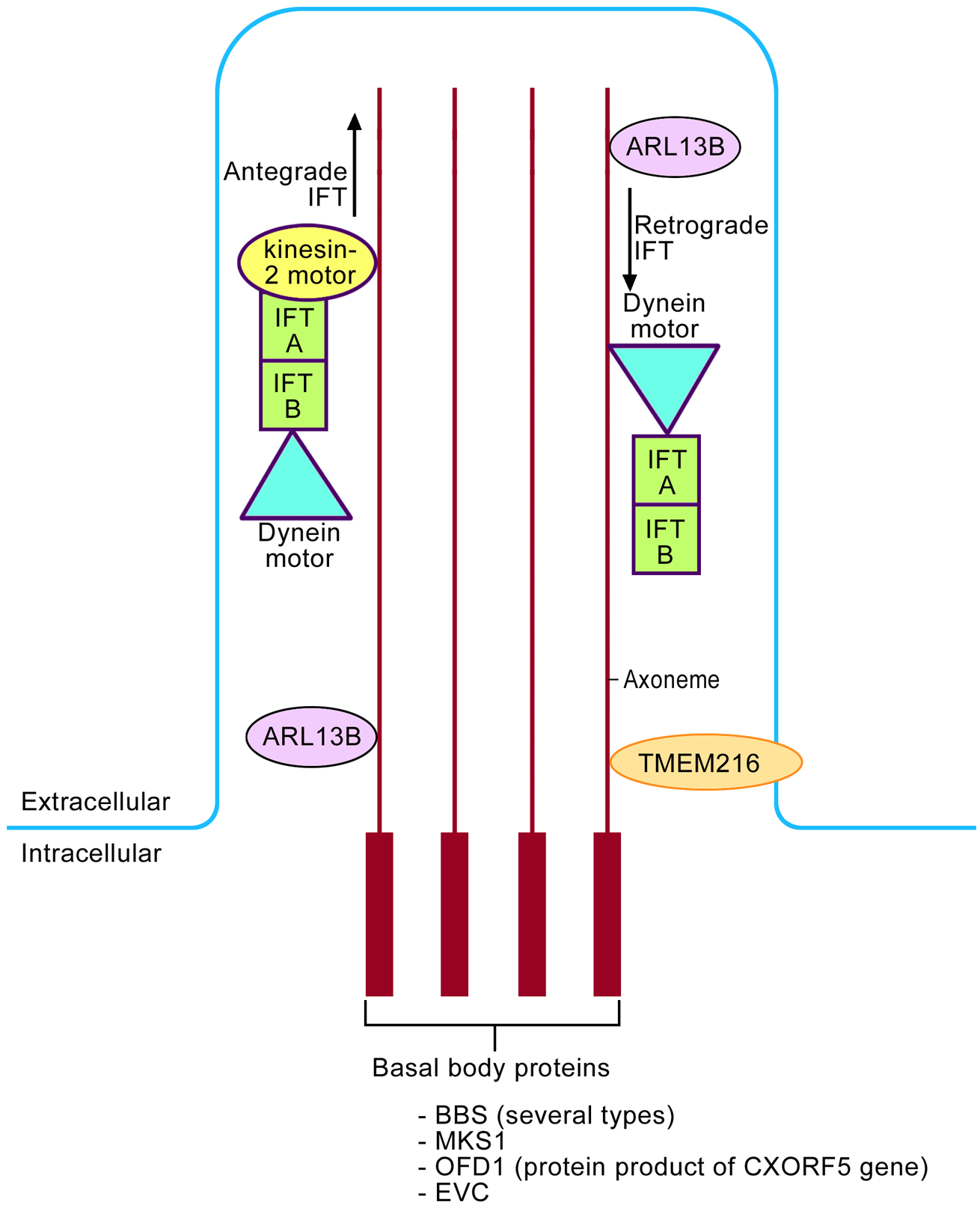
The basic structure of the primary cilium and how it is involved in the intraflagellar transport (IFT). The basic structure includes the basal body proteins and axoneme. Antegrade IFT is mediated via the kinesin-2 motor and IFT A/B protein complexes. The dynein motor is mainly involved in retrograde IFT. ARL13B, a small GTPase, localizes to the cilia and is required for axoneme structure. TMEM 216 (transmembrane protein 216) also localizes to the cilium.
Figure 3 shows the processing of Gli3 within the cilium via the Shh ligand and its receptor, patched 1 (PTCH1). PTCH1 is located at the base of the ciliary membrane which blocks smoothened (SMO). Therefore, Gli3 cannot be processed in the anterior limb bud where there is no Shh, and Gli3-R predominates. In the posterior limb bud, Shh binds to PTCH1. This results in movement of the receptor away from the base of the cilium allowing entry of SMO to the ciliary membrane. Hence Gli3 is processed (via the kinesin KIF7) through the IFT (Figure 2) into the active form of Gli3-A. This will provide the normal balanced gradients between Gli3-R anteriorly and Gli3-A posteriorly, as shown in Figure 1.
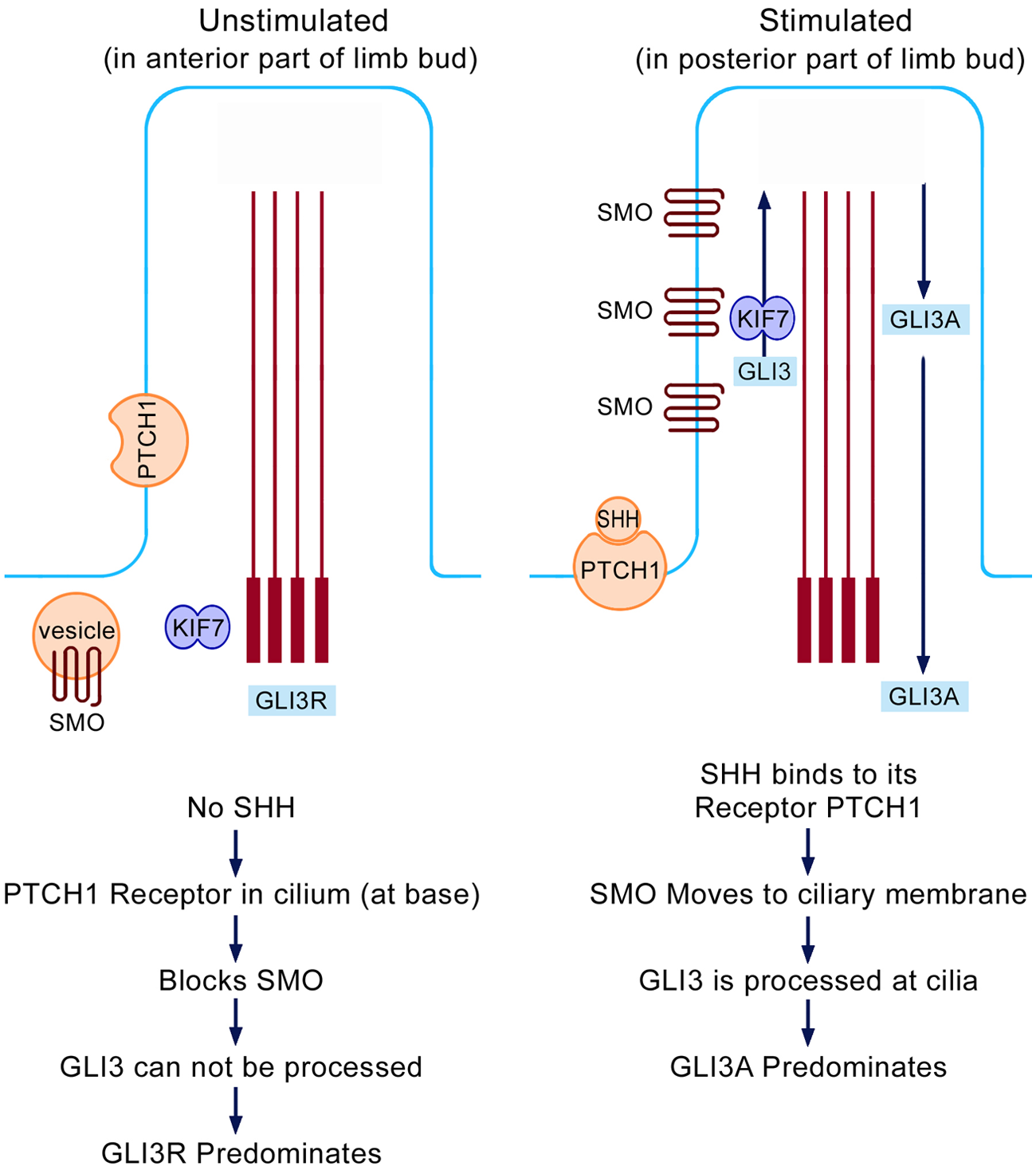
Processing of Shh and Gli3 within the primary cilium. The unstimulated condition is seen in the anterior limb bud (no Shh activity) while the stimulated condition is seen in the posterior limb bud (Shh is located in the posterior mesenchyme).
Classification of syndromes associated with UP
Syndromes associated with UP may be classified into three main groups: ciliopathy syndromes that are caused by gene mutations related to the structure of the cilia or the IFT; mutations in the GLI3 gene leading to truncations of the Gli3 protein; and chromosomal duplication syndromes in which UP is a prominent feature, such as Patau syndrome (Trisomy 13). The latter group of syndromes will not be discussed here because the exact effect of chromosomal duplication is poorly understood.
Ciliopathy syndromes
Ciliopathy syndromes are summarized in Table 1. Although different syndromes have different phenotypes, they all share the feature of UP. The causative gene mutations in these syndromes interfere with either the normal structure of the cilia or structure of proteins involved in the IFT of the Gli3 protein. As a result, the production of Gli3-A is relatively decreased leading to a relative increase in the Gli3-R. Mutations of the MKKS gene (also known as BBS6) cause McKusick-Kaufman syndrome and one type of Bradet-Biedl syndrome. MKKS has a chaperonin-like function. Therefore, mutations of MKKS will result in the inability of the MKKS putative chaperonin to maintain protein integrity in the cilia (Slavotinek et al., 2000). The Bradet-Biedl syndrome phenotype is also caused by mutations in several BBS genes, which encode several basal body protein-BBSome components such as BBSomes 1,2,4,5 and 7. Mutations of the EVC gene, which encodes basal body protein-skeletal specific (EVC) proteins, cause Ellis-Van Creveld and Curry-Hall syndromes. A mutation of the MKS1 gene (which encodes the MKS1 basal body protein) causes Meckel-Gruber syndrome type 1. Finally, the CXorf5 (chromosome X open reading frame 5) gene encodes a basal body protein known as OFD1 (oro-facial-digital type 1) protein. Mutations of CXorf5 cause oral facial digital syndrome type 1, Simpson-Golabi-Behmel syndrome type 2, and one type of Joubert syndrome. In all of the above mutations, the normal basal body protein structure is disturbed, leading to defective ciliary function and hence reduced Gli3 processing.
Ciliopathy syndromes with ulnar polydactyly (UP).
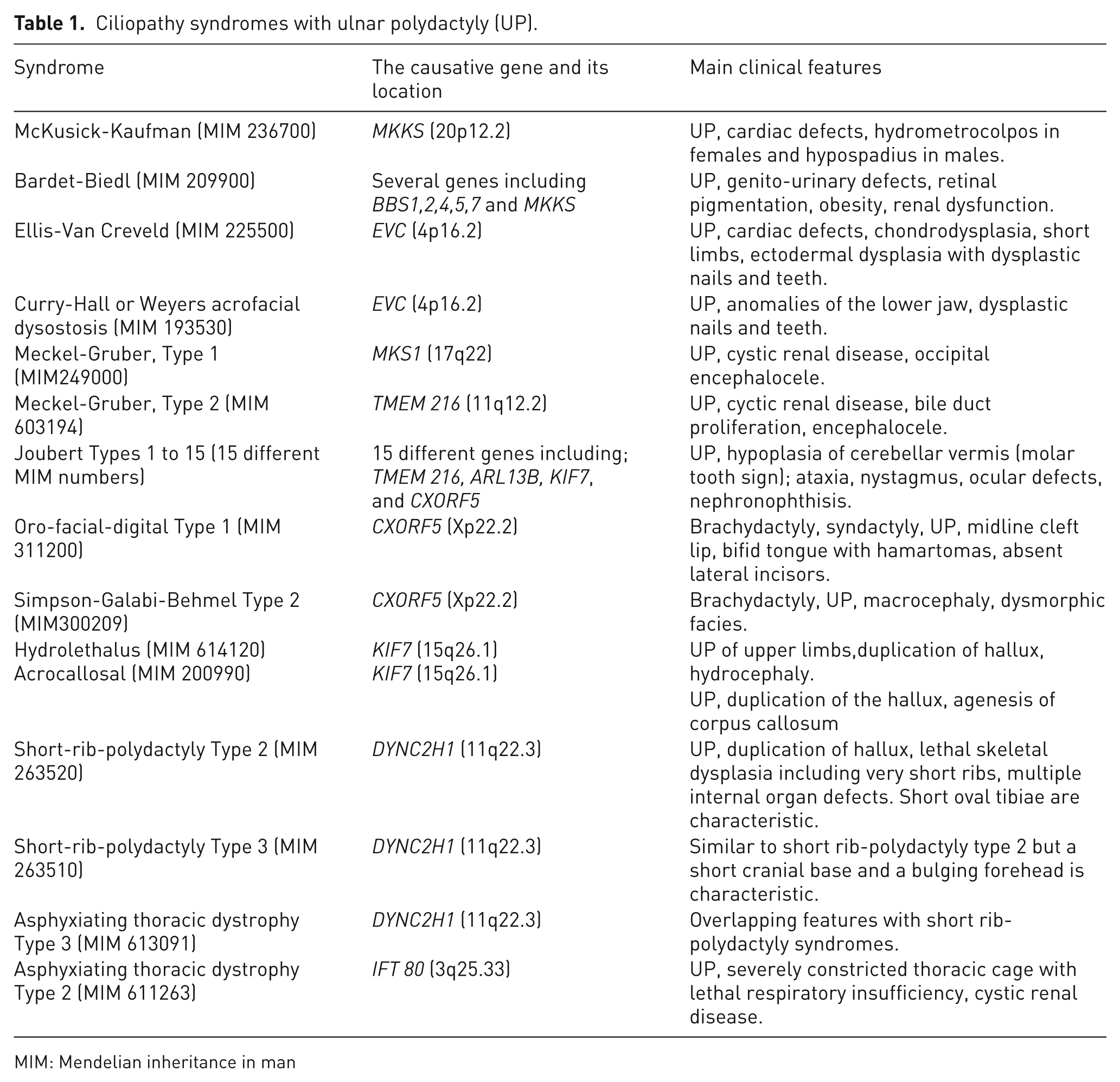
MIM: Mendelian inheritance in man
The remaining ciliopathy syndromes (Table 1) are caused by mutations of genes encoding proteins involved in the IFT of Gli3, such as TMEM216, ARL13B, KIF7, DYNC2H1 and IFT80 (IFT80 protein is part of IFT B protein complex) (Table 1 and Figure 2). Once again, the genetic defects will disturb the processing of Gli3 and hence reduce the production of its active form.
GLI3 mutation syndromes
The GLI3 gene is a member of the zinc finger gene family related to Krüppel (a gene that regulates development in drosophila). In humans, the gene is located at 7p14.1. GLI3 mutation syndromes have UP in their phenotypes and these are summarized in Table 2. The Gli3 protein may be divided into three parts (Figure 4). The part towards the N-terminal contains the zinc finger domain (ZFD). Mutations in the GLI3 gene that predict truncation of the Gli3 protein before or within the ZFD result in Greig cephalopolysyndactyly syndrome (GCPS). These mutations are considered as null mutations (haploinsufficiency) caused by loss of the zinc finger DNA-binding domain (Biesecker, 1997; Johnston et al., 2010). Patients with Pallister-Hall syndrome have protein truncation after the ZFD but before the transactivation domains (i.e. the truncation is within the middle part of the Gli3 protein). This results in over-abundance of Gli3-R (Johnston et al., 2005). Finally, GLI3 mutations that predict truncations in the carboxyterminal part of the Gli3 protein will cause a variable degree of loss of the transactivation domain of GLI3. This results in a variable degree of abundance Gli3-R; and the resulting phenotype is either GCPS or isolated UP types A/B (Kalff-Suske et al., 1999; Radakhrishna et al., 1997). It should be noted that the structure of the cilia and the IFT basic functions are all normal in this group of syndromes with GLI3 gene mutations. Instead, the abundance of Gli3-R is owing to truncation of the Gli3 protein itself, leading to a variable degree of loss of the active form of Gli3.
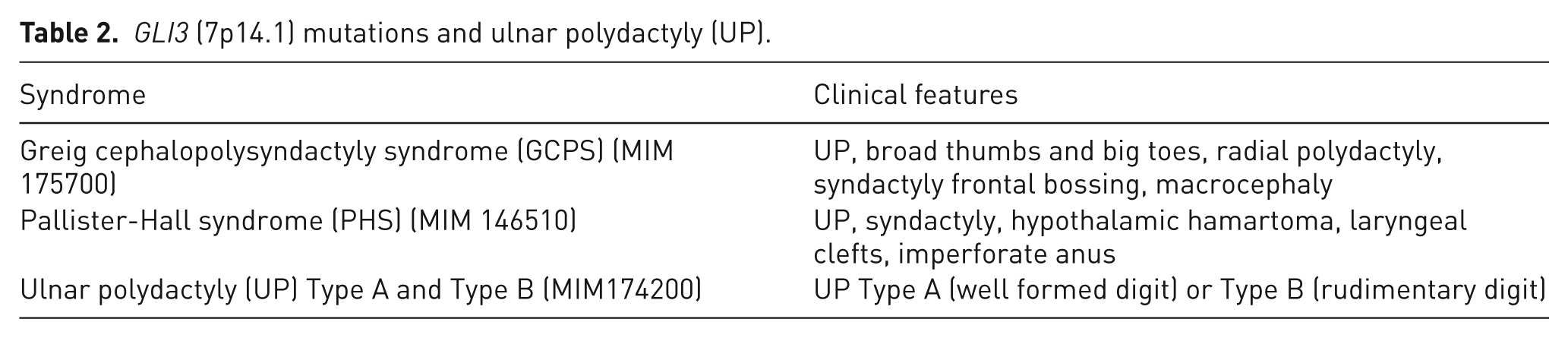
GLI3 (7p14.1) mutations and ulnar polydactyly (UP).
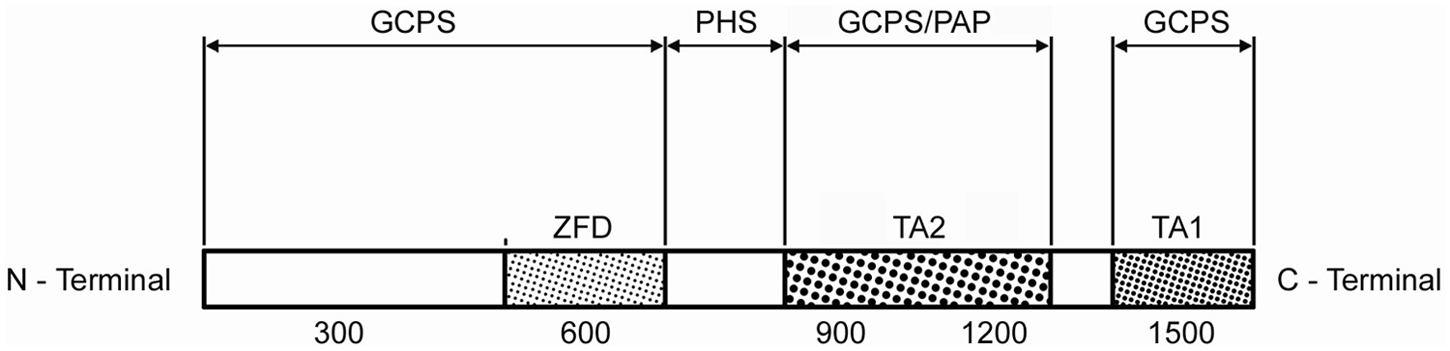
The three parts of the GLI3 protein. See text for details.
Finally, it is important to note that patients with GCPS and GLI3 mutations may have simultaneous ulnar and radial polydactyly (Table 1). The reason for this may be through the well-known interactions between Gli3 and HOXD. Chen et al. (2004) have shown Gli3-Hoxd12 interactions experimentally, and defects in Hoxd12 in mice are known to result in radial polydactyly (Kmita et al., 2005).
Conclusion
Although UP is a common congenital hand anomaly, its exact pathogenesis remains unknown. There is skewing of the normal balance of Gli3A-Gli3R (with predominance of Gli3R) within the embryonic ‘hand plate’ in syndromes featuring UP. This opens new insights and may be a guide for future experimental research on the topic.
Footnotes
Funding
This work was supported by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University, Riyadh, Saudi Arabia.
Conflict of interests
None declared.