
Abstract

Dear Editor,
There has been increasing evidence associating the development of atraumatic, multifocal osteonecrosis to hypercoagulable states (Baltzer et al., 2016; Woratanarat et al., 2014). Congenital hypercoagulability, such as Factor V Leiden (FVL), resulting in thrombosis may compromise bone vascularization to increase the risk of osteonecrosis. Herein, we report a rare case of bilateral Kienböck’s disease in a 14-year-old, right-hand dominant male. He was diagnosed with heterozygous FVL and the Kienböck’s disease subsequently treated with 4, 5 extensor compartment supraretinacular artery (ECA) bone grafts.
The patient developed pain over a 3-month period. There was swelling along the dorsal side of the right wrist compared with the left, with decreased range of motion and grip strength without redness or warmth. There was no history of injury, and also no history of deep venous thrombosis or bleeding disorders. His family history consisted of a maternal history of rheumatoid arthritis and seizure-type disorder. Radiographs of the right wrist revealed sclerosis and mild radial flattening of the lunate with a negative ulnar variance. At around the same time, the patient developed a similar left wrist pain. There was mild lunate sclerosis with a negative ulnar variance noted on X-ray film. MRI of the right wrist revealed a nondisplaced fracture of the dorsal third of the lunate with diffuse marrow oedema and bony irregularity, suggestive of impaired lunate vascularity. On the left side, MRI revealed decreased craniocaudal diameter on the radial aspect of the lunate, with patchy marrow lunate oedema (Figure 1). The results were consistent for stage IIIA Kienböck’s disease on the right and stage II on the left side.
Bilateral wrist MRI of the patient, demonstrating stage II on the left and stage IIIA on the right side.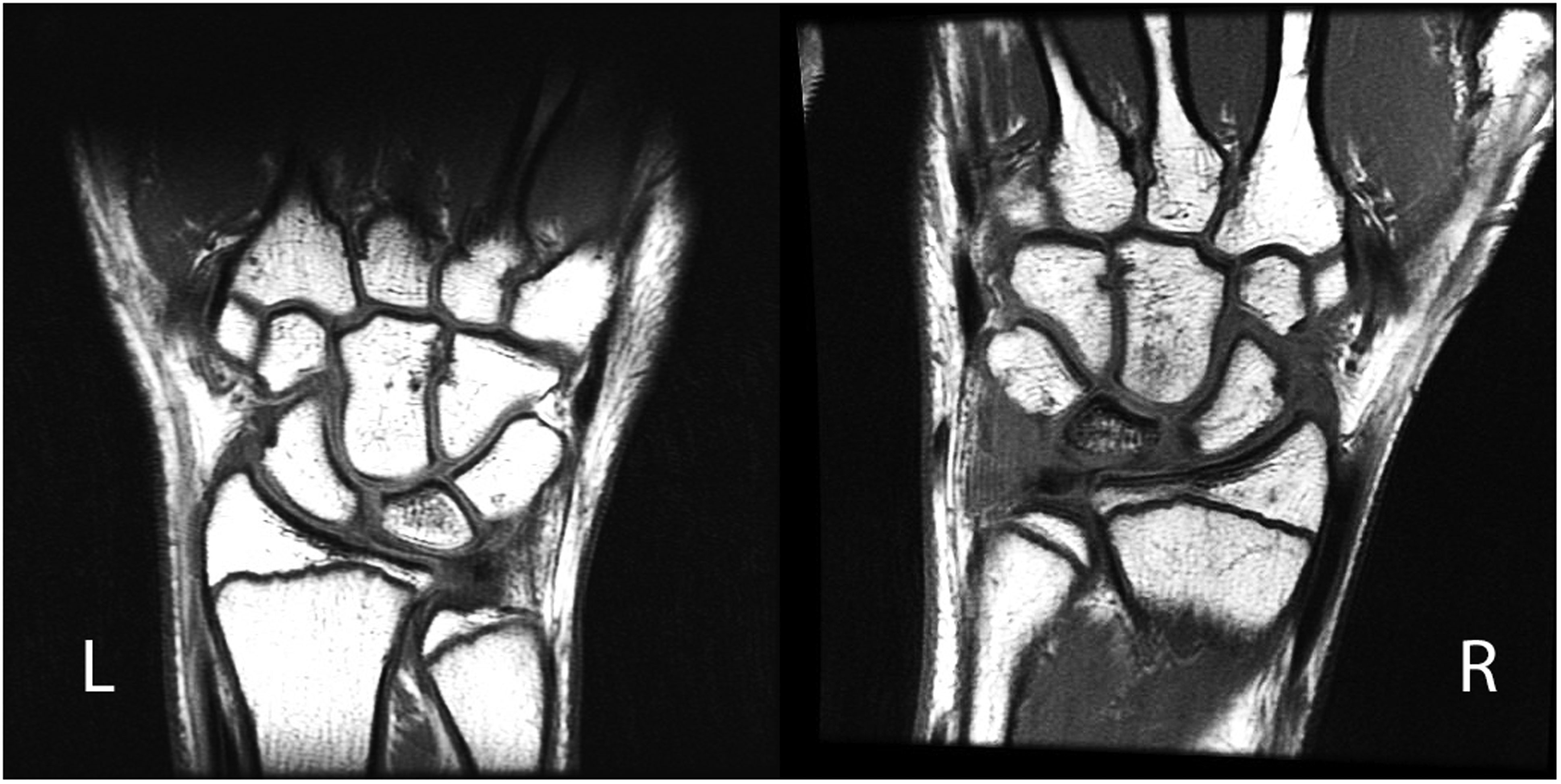
Previous experience had alerted us to the possibility of hypercoagulopathy (Baltzer et al., 2016) and a workup was performed, which included protein C and S activities, antithrombin activity, soluble fibrin monomer, D-dimer, fibrinogen, thrombin time, activated partial thromboplastin time (APTT), coagulation time, prothrombin G20210A mutation, autoantibodies, complete blood count with differentials, and FVL. The patient had a low APCRV (Activated protein C resistance V, 1.6), elevated PT (13.6 seconds), and heterozygous mutation for FVL.
The patient underwent reconstruction of the right lunate with a 4 + 5 ECA graft. Subsequently 3 months later, the left lunate also received the same reconstruction. The postoperative course was uneventful. At the latest follow-up (1-year right wrist reconstruction), the patient reported no pain or paraesthesia of the wrists and no restrictions to daily activities, with a reasonable and symmetric range of motion in bilateral wrists and all fingers. Wrist MRI demonstrated improvement of the lunate bone marrow signal with decreased effusion and synovitis that were consistent with ongoing healing. The patient was followed by the haematologists, although no formal anti-coagulation was recommended.
Kienböck’s disease in children is rare. A group reviewed radiographs of 51,071 patients over an 11-year period with a prevalence of 0.27% in their population (van Leeuwen et al., 2016). Currently, no consensus has been reached with regards to the aetiology of Kienböck’s disease. Besides anatomical variations, biological factors, such as immunological, microbiological, and genetic factors, have been postulated to play important roles in the initiation of the disease to induce reactive vascular aberrations in the lunate’s subchondral vasculature (Van Alphen et al., 2016). The at-risk lunate can then be further aggravated by mechanical factors resulting in osteonecrosis of the bone (Van Alphen et al., 2016). Many studies have reported associations between the developments of multifocal osteonecrosis to coagulopathies. FVL is the most common, as demonstrated by a recent meta-analysis that showed strong correlation between genetic hypercoagulability to Legg–Calve–Perthes disease (LCPD) (Woratanarat et al., 2014).
Idiopathic bilateral Kienböck’s disease in a child raises questions regarding its aetiology; it has been suggested that paediatric lunate avascular necrosis should be regarded as a separate disorder, in the same way that femoral head avascular necrosis in adults and children (LCPD) should be distinguished, as the aetiology and treatments are different (Irisarri et al., 2010). Given the rarity of Kienböck’s disease in children and the scarcity of reports, it is difficult to draw firm conclusions associating the development of Kienböck’s disease to hypercoagulability. However, the connection cannot be overlooked given the strong association between LCPD and genetic hypercoagulability (Woratanarat et al., 2014). Thromboembolic events from hypercoagulability may just be one of the more common factors in children, and the adult pool of factors may be very different.
Bilateral Kienböck’s disease is extremely rare in children. Hereditary thrombophilic disorders may predispose the lunate to avascular necrosis. A high suspicion for FVL is warranted for children with bilateral Kienböck’s disease.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.