
Abstract
This study reports adverse events after adjuvant radiotherapy in patients with Dupuytren’s disease. Patients for planned limited fasciectomy, collagenase injection or percutaneous needle fasciotomy were enrolled in a randomized controlled trial. Sixty patients were randomized to observation or radiotherapy (30 Gy, 10 fractions) starting within 4 days of the procedure. During a follow-up period of up to 4 years, 114 adverse events were reported. Of 102 (89%) mild adverse events, 45 (44.1%) were attributed to radiotherapy. Of the 12 moderate to severe, three were attributed to radiotherapy. By 6 months, most moderate to severe adverse events had resolved. Between 12 and 36 month follow-ups, the only persistent adverse event attributed to radiotherapy was reduced sweating. In this trial of adjuvant radiotherapy for Dupuytren’s disease, adverse events are generally mild and self-limiting, suggesting that adjuvant radiotherapy is well tolerated.
Introduction
Dupuytren’s disease (DD) is a benign fibroproliferative disorder characterized by the development of nodules and cords in the palm. The typical pathological feature of DD is abnormal myofibroblastic growth in the hand, producing excess type 3 collagen (Broekstra et al., 2022). All therapeutic interventions have an associated recurrence rate, mandating exploration of adjuvant treatments to improve longer term disease control (van Rijssen et al., 2012).
Radiation therapy (RT) is a cytotoxic, non-invasive intervention that uses high-energy photons or electrons targeted to a specific anatomical region. Although often used for malignant disease, it is also an established and recognized treatment in the management of benign hyperproliferative disorders. Low- and moderate-dose RT targets abnormally rapidly proliferating cells, producing a mitotic arrest (Seegenschmiedt et al., 2015; The Royal College of Radiologists, 2023).
Several factors providing a rationale for the role of adjuvant RT in DD have been described (Lambi et al., 2023). In early DD, the proliferating fibroblast is an ideal target for RT, being localized, radiosensitive and active in the cell cycle. Furthermore, other conditions, such as keloid scarring and heterotopic ossification, caused by similar processes have evidence to support the use of RT in reducing relapse (Milakovic et al., 2015; Seegenschmiedt et al., 2015; Wolfram et al., 2009).
The role of RT in early DD has been investigated (Adamietz et al., 2001; Betz et al., 2010; Keilholz et al., 1996). These uncontrolled studies are limited by the unpredictable natural history of the condition and lack of randomized data, but support the concept of prophylactic RT. Despite the limitations of this evidence, some guidelines advocate for its use (Seegenschmiedt et al., 2015). This remains controversial, with a recent systematic review finding weak evidence for RT as a preventative strategy for DD and calling for a randomized trial to address this issue (Kadhum et al., 2017).
This study aimed to report early adverse events (AEs) for adjuvant RT for patients participating in a randomized trial assessing the role of radiation therapy in DD.
Methods
Study overview and protocol
The Dupuytren’s disease Evaluation of Preventative or Adjuvant Radiation Therapy (DEPART) trial is an open-label, multicentre, randomized, controlled trial assessing the role of radiotherapy in DD. The study aims to recruit patients across two components: preventative and adjuvant. This report focuses on the adjuvant component, which has three groups: limited fasciectomy (LF), percutaneous needle fasciotomy (PNF) and collagenase injections (CI). In all groups, participants were randomized to either a control arm of observation or an experimental arm of radiotherapy. The Trial is prospectively registered (ACTRN12618000951257) and has Human Ethics Research Approval (Bellberry 2018-02-134). GenesisCare financially sponsored this investigator-initiated and clinically managed trial.
Recruitment
The DEPART trial began recruitment in September 2018. In January 2022, the trial management committee recommended halted recruitment to the adjuvant component for two reasons: collagenase was withdrawn from the Australian market in 2021, challenging recruitment to the CI group; and there was a newly developed consensus on the definition of recurrence (Felici et al., 2014). The trial committee then enacted a protocol amendment to include only PNF patients in the adjuvant arm and, using the new recurrence definition, restarted patient recruitment in November 2023. Data from the initial participants (2018–2022) were used to report on adverse events.
Inclusion and exclusion criteria
Inclusion criteria were patients aged over 30 years with an estimated life expectancy of more than 5 years, who agreed to participate in at least 5 years of follow-up and were able to provide written informed consent. Patients for whom local treatment with LF, PNF or CI of flexion contracture(s) was planned were eligible for the adjuvant component. If there was doubt about the clinical diagnosis of DD, pathological confirmation was recommended.
Patients were excluded from the study if they had received prior radiotherapy to the hand, had a history of radiosensitivity disorders (such as scleroderma, ataxia telangiectasia or li fraumeni), were pregnant or could not start radiotherapy within 4 days of surgery because of a significant complication (for example tendon rupture or infection), or if their hand could not be positioned flat on the radiotherapy table.
Randomization and treatment
Patients in the adjuvant group were randomized to either observation or adjuvant radiotherapy. Randomization was performed using a stratified random permuted block design with LF, PNF and CI as stratification factors.
All surgeons contributing to this trial had at least level 3 expertise (Tang and Giddins, 2016). Fasciectomy included limited fasciectomy, dermofasciectomy or other fascial debulking procedure. Participants randomized to radiotherapy had to have commenced their therapy no more than four calendar days after the intervention. The rationale for this timing was based on the heterotopic ossification literature and the knowledge that the acute inflammatory reaction mediators with new collagen deposition peaks within 1–4 days (The Royal College of Radiologists, 2023; Wolfram et al., 2009).
The radiotherapy treatment dose prescribed was 30 Gy in 10 fractions. The target area was defined as the regions of involved DD, including any scar/wounds, with a 1–2 cm margin to allow sufficient coverage whilst avoiding any exit dose through the nail matrix (Figure 1). Radiotherapy was delivered at three to five fractions per week. After the first five fractions, a break of 4–12 weeks was taken before completion of the remaining fractions. Centralized radiotherapy quality assurance for each centre was assessed to ensure compliance with the treatment protocol.
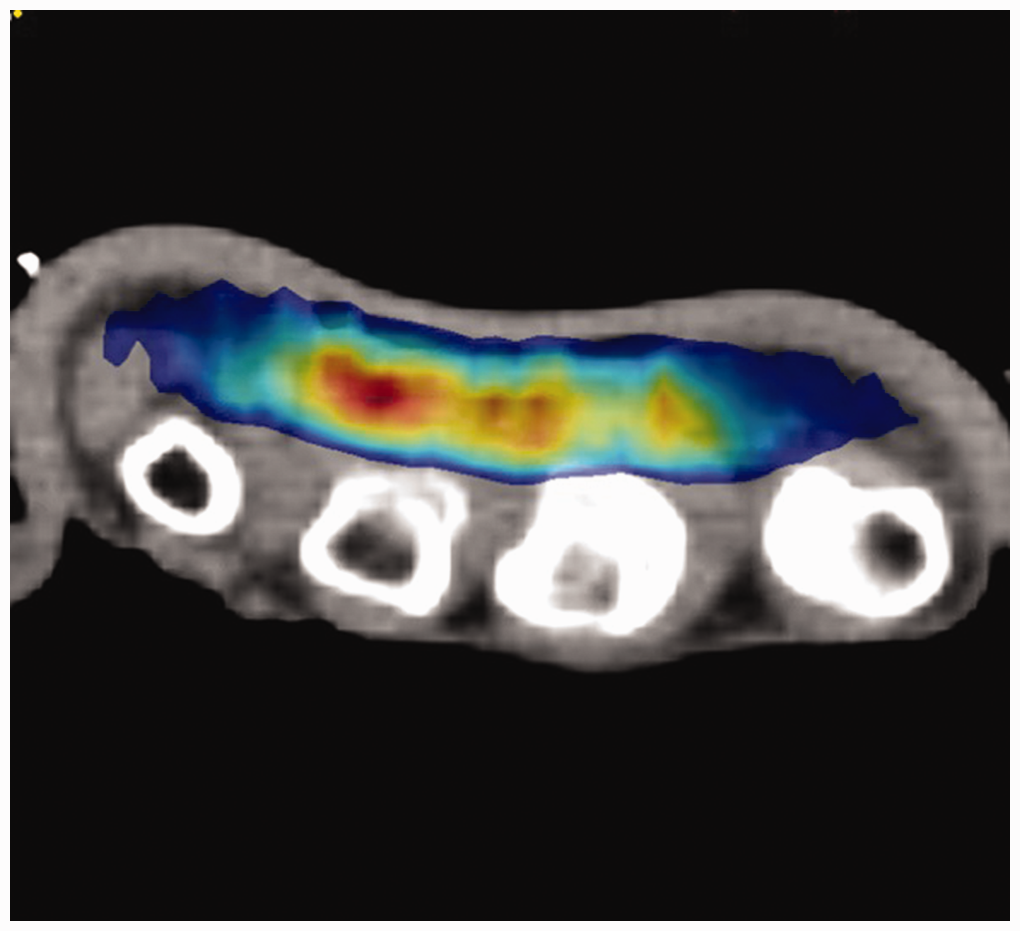
Hand radiotherapy dose distribution achieved using 6 MeV electrons and a 5 mm customized bolus shaped to the palmar/proximal digital fascia. Note that most of the dose is limited to the subcutaneous structures of the palmar surface.
Patients randomized to radiotherapy underwent baseline and review assessments. All trial participants were independently reviewed by a certified hand therapist after the procedure, at 6 months, 12 months, annually up to 60 months and then biannually up to 104 months.
Adverse events
Adverse events were categorized using the Common Terminology Criteria for Adverse Events (CTCAE) scoring system v4.03 with toxicity graded using a scale from 0 to 5, where 0 is no toxicity, 1 is mild, 2 is moderate, 3 is severe, 4 is potentially life threatening and 5 is death. In general, a grade 1 AE is noticeable but does not require intervention. A grade 2 AE would require an outpatient intervention (e.g. medication) and a grade 3 AE would require a hospital admission. For subjects randomized to no radiotherapy, all AEs were attributed to the surgical intervention. For the radiotherapy arm, an AE localized to the wound was attributed to the initial intervention, whereas more general effects were attributed to radiotherapy. Idiosyncratic effects such as reduced sweating were attributed to the radiotherapy. Patient reported outcomes were recorded using the visual analogue scale for pain, the Unité Rhumatologique des Affections de la Main for DD and the QuickDASH. These outcomes are not reported in this paper.
Statistics
Descriptive statistics are provided for comparative purposes between the two arms (observation and RT) and across the three adjuvant groups (LF, PNF and CI). Continuous data were assessed for normality and accordingly are reported as mean or median with range. The χ2 test or Fischer’s exact test (when any cell count was <5) were used to assess differences in categorical variables between groups. Results were considered statistically significant at a p-value of less than 0.05.
Results
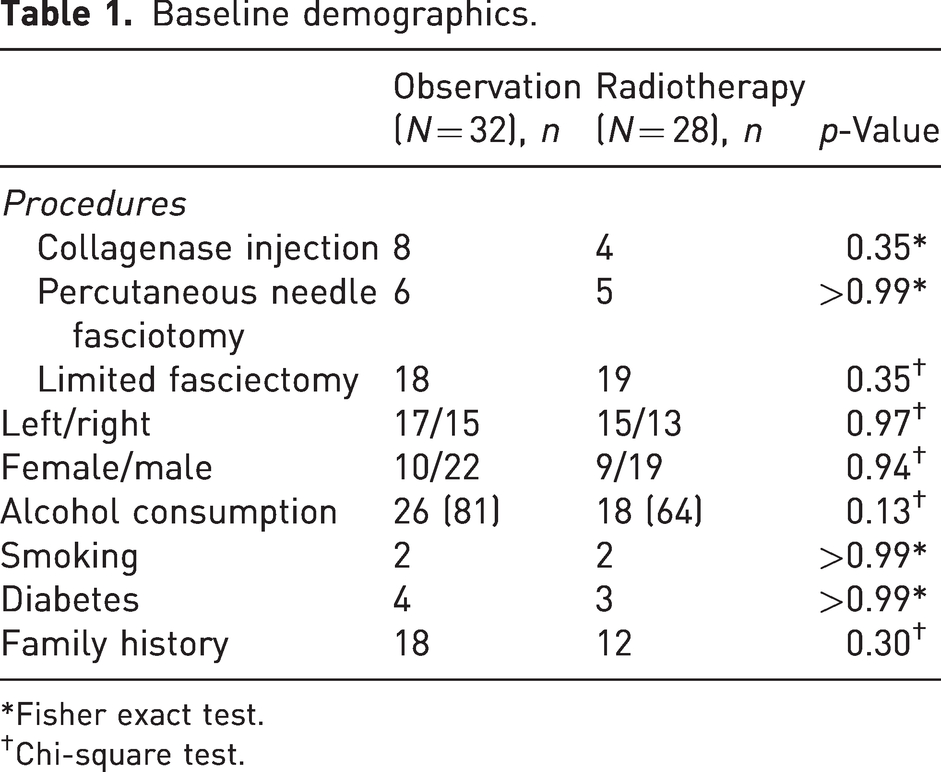
Sixty patients were enrolled on the adjuvant arm of the DEPART trial from six Australian centres. The mean age was 63 (range 37–78) and the median follow-up was 12 months (range 1.5–48). Baseline demographics and clinical characteristics are outlined in Table 1.
Baseline demographics.
*Fisher exact test.
Chi-square test.
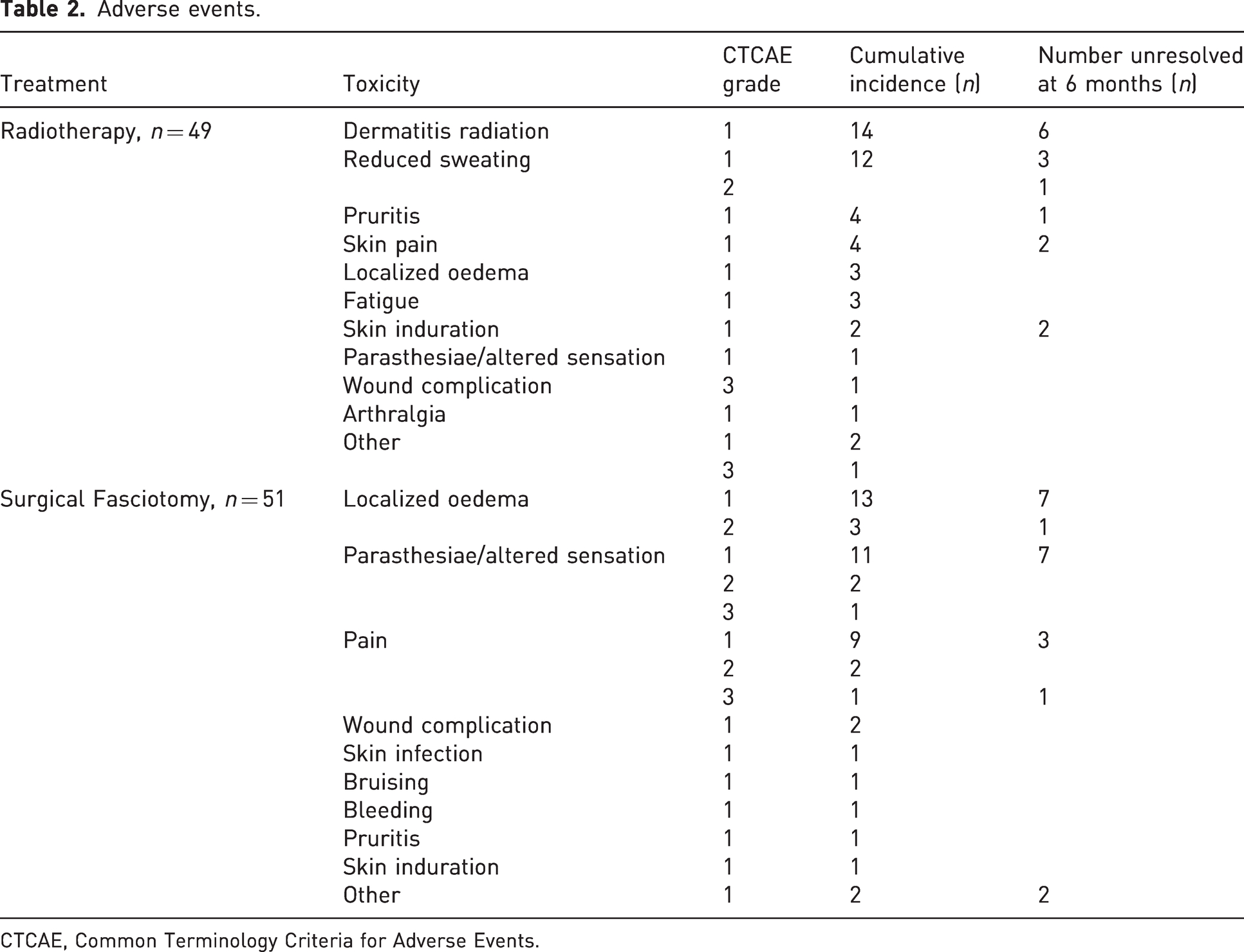
Thirty-nine patients reported 114 AEs based on CTCAE scoring. Of this group, the median number of AEs per hand was 2 (range 1–8). The majority (102) were mild (grade 1) and did not require intervention. The AEs are outlined in Table 2. Figure 2 outlines the prevalence of AEs for RT and LF at follow up.
Adverse events.
CTCAE, Common Terminology Criteria for Adverse Events.
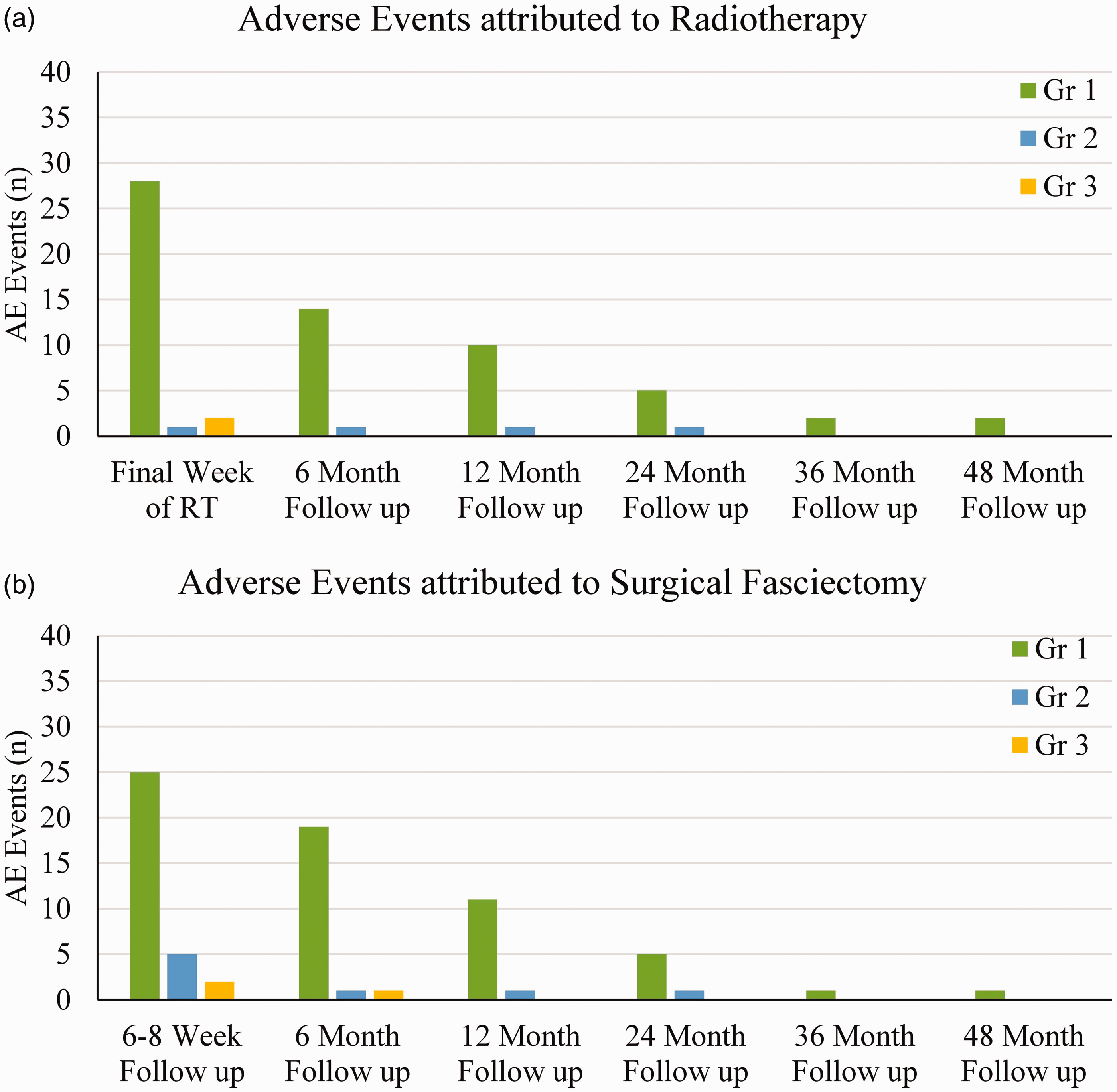
Prevalence of adverse events attributed to (a) radiotherapy and (b) fasciectomy. Note that in both cases, the majority of events are mild, peak early and are largely self-limiting.
Six patients experienced grade 2 CTCAE with eight AEs recorded, including localized oedema, pain, paresthesia/altered sensation and reduced sweating of the palm. Three participants experienced a grade 3 CTCAE with four AEs. The first was a patient in the LF observation cohort who experienced significant pain at the 6–8 week follow-up review which had not resolved at 6 months follow-up. The second patient, also in the LF observation cohort, experienced parasthesiae/altered sensation at the 6–8 week follow-up review, which resolved at 6 months follow-up. The last was a patient in the LF adjuvant radiotherapy cohort who developed a grade 3 AE of wound dehiscence with bleeding before the first fraction of radiotherapy, attributed to forced extension of the hand. Radiotherapy was delayed for this patient until after the wound had healed. This event prompted a protocol revision, excluding patients who were unable to position their hand flat.
Radiotherapy was interrupted in two patients, the first being the patient who experienced grade 3 wound dehiscence and the other a patient with a skin tear in the CI/observation group.
The only unrecovered late radiotherapy AEs at the last follow-up were reduced sweating of the palm in seven patients, and one patient noticed reduced pigmentation of the skin. There were 21 patients with over 12 months of follow-up in the radiotherapy cohort and 22 patients in the observation cohort. At the 12 month prevalence time-point, the G1 and higher AE were not significant between the two cohorts (p = 0.38). With a median follow-up of 12 months, the cumulative G2 or higher AE worst incidence per patient was not significantly different between the two cohorts (Table 3). Figure 3 outlines the AE prevalence for the adjuvant radiotherapy cohort and the observation cohort, respectively.
Worst adverse event grade toxicity reported per patient.
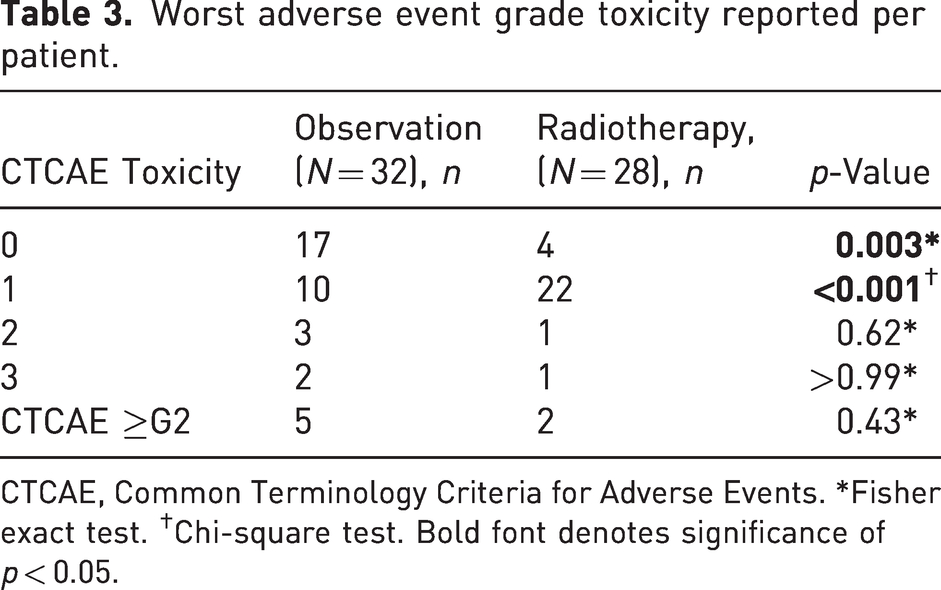
CTCAE, Common Terminology Criteria for Adverse Events. *Fisher exact test. †Chi-square test. Bold font denotes significance of p < 0.05.
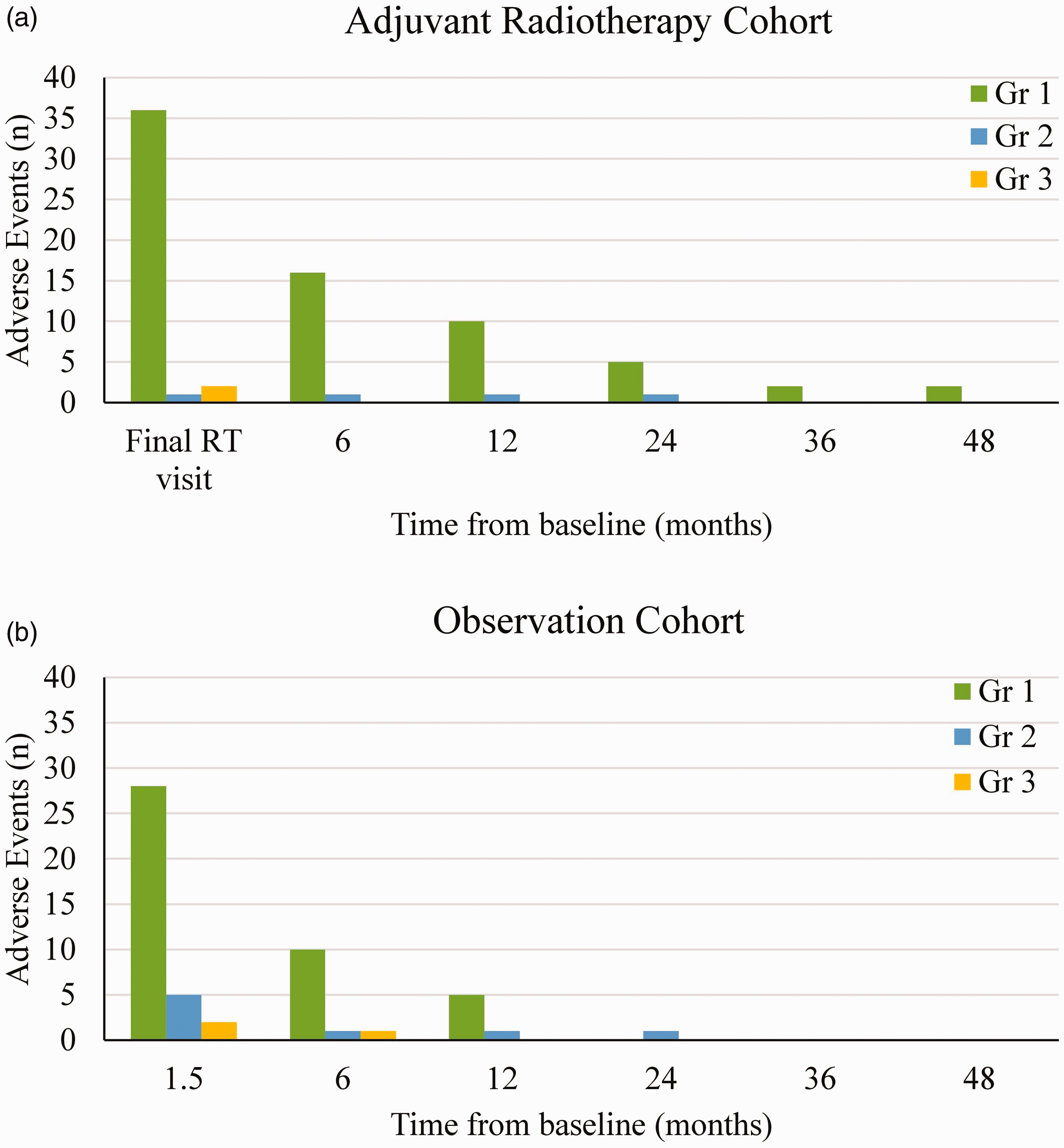
Prevalence of adverse events for (a) the adjuvant radiotherapy cohort and (b) the observation cohort.
Discussion
This study has demonstrated that adjuvant radiotherapy in the management of Dupuytren’s disease is feasible and safe with early to mid-term follow-up. The majority of adverse events were mild, short-term erythema. Reduced sweating, although mild, tended to persist. Wound complications were rare with adjuvant RT and similar to those seen after fasciectomy alone.
The rationale for adjuvant RT in DD is to limit the proliferation of repopulating fibroblasts to reduce the risk of recurrence. In this study, there was no observed delay in wound healing, probably from the relatively low dose of RT used and the excellent blood supply to the palm. Randomized trials of adjuvant RT to prevention recurrence of heterotopic ossification have proven this to be a safe and effective method (Milakovic et al., 2015; Pakos and Ioannidis, 2004). Lower quality data suggest a similar effect after keloid scar revision by limiting fibroblast proliferation (Seegenschmiedt et al., 2015; Wolfram et al., 2009). There are very limited, uncontrolled data for adjuvant RT in DD. A small German series of brachytherapy immediately after LF in six patients (13 digits) reported no toxicity and no recurrence at a mean follow-up of 14.1 months (Ciernik et al., 2021). The biological rationale and evidence from other disease processes, alongside our study findings, support the need for further research into adjuvant RT for DD.
There is ongoing concern about the risk of malignancies after the use of RT for DD. By limiting treatment to people over the age of 30 to a peripheral location, using a simple RT technique and a low RT dose, we aim to achieve rates similar to those estimated by the Royal College of Radiologists (<1:5000) (The Royal College of Radiologists, 2023). To our knowledge, only one case of an RT-induced malignancy has been reported in a patient treated for hyperhidrosis as a teenager who developed a cutaneous squamous cell carcinoma 40 years later (Campanelli and Lubbe, 2019). No sarcomas have been reported, with the majority of evidence suggesting that RT is most likely to increase the incidence of common tumours in the irradiated area (Bagshaw et al., 2022). This all reinforces the need for high-quality evidence of advantage before adjuvant RT is advocated as a standard approach and why the small risk of malignancy needs to be part of any informed consent process before such treatment.
This study is strengthened by the prospective, independently collected data. There are, however, weaknesses of the study. The study has only 60 patients with three subcategories, precluding any useful separate analysis. A further weakness is the lack of patient-reported outcome data, including quality of life, although this data will be reported in due course. The study is weakened by the challenges of collagenase withdrawal from the market, slow recruitment and the redefinition of recurrence, which ultimately led to recruitment being ceased with a major protocol amendment in response to these circumstances (Felici et al., 2014).
Although there are advocates for the use of RT in the prevention of DD and weak evidence for its use after intervention, the current literature suggests that a stronger evidence base is necessary before RT can be routinely recommended in either setting (Kadhum et al., 2017; Seegenschmiedt et al., 2015). The DEPART trial should eventually provide important data on any potential benefit from RT as a preventative approach. The adjuvant component has now been relaunched, focusing on the PNF cohort given the high risk of recurrence at over 5 years in this group (van Rijssen et al., 2012). Trial patients now undergo RT 6–12 weeks after PNF to maximize healing and allow reliable baseline measurements to be obtained (Felici et al., 2014).
This study suggests that RT after intervention is safe, with minimal long-term sequelae. Importantly, there has been no significant impact on wound healing after surgery, supporting its use as a safe adjuvant treatment. Further research is required to assess the acute and chronic AEs as well as the efficacy of RT as an adjuvant treatment to reduce recurrence after intervention.
Footnotes
Acknowledgement
The authors appreciate the contribution of other DEPART Clinical Trial Collaboration members in the study design, recruitment and data collection: Dion Sandoz, Joseph Bucci, Anneke de Haan, David Dilley, Philippa Ell, Kathryn Hogan, Katherine Neville, Warren Rozen, Sandy Sampaio, Joshua Sappiatzer and Roel JHM Steenbakkers.
Declaration of conflicting interests
GenesisCare is a commercial entity which includes radiotherapy treatment and which funded the conduct of this study. All analysis, interpretations, and write up was independent of GenesisCare. JM owns stock in GenesisCare and has received support from GenesisCare for the conduct of the research. Other authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
GenesisCare financially sponsored this investigator-initiated and clinical managed trial.
Ethical approval
Ethical approval for this study was obtained from Bellberry Human Research Ethics Committee (2018-02-134).
Informed consent
Written informed consent was obtained from all subjects before the study.