
Abstract
Background:
Birt-Hogg-Dubé (BHD) syndrome is an autosomal dominant disorder caused by folliculin (FLCN) gene mutations, with ethnically heterogeneous mutational spectra. Current management is primarily supportive and lacks curative therapies.
Objectives:
This study aimed to characterize the clinical features and genetic variants in two distinct Chinese families with BHD syndrome and to preliminarily explore the feasibility of an mRNA-based protein replacement approach.
Design:
A family-based prospective cohort study was conducted to identify FLCN mutations in two BHD families carrying rare FLCN variants enrolled in 2023.
Methods:
Whole-exome sequencing (WES) identified candidate mutations in probands, which were validated by Sanger sequencing (p.W376R and p.Q44*). Structural/functional impacts of p.W376R were analyzed via bioinformatics and qPCR. HEK293T cells were transfected with empty vector, wild-type FLCN, or mutant plasmids (p.W376R and p.Q44*), followed by co-transfection with or without synthetic FLCN mRNA. FLCN expression and mTORC1 signaling were assessed.
Results:
Affected members in both families predominantly presented with respiratory symptoms, lacking typical skin lesions and kidney tumors. WES identified the FLCN variants p.W376R (previously classified as a Variant of Uncertain Significance, VUS) and a novel nonsense mutation p.Q44*. Genotype–phenotype co-segregation was confirmed for p.W376R. In vitro analyses demonstrated that both mutations reduced FLCN protein expression, leading to mTORC1 hyperactivation. Exogenous FLCN mRNA delivery rescued functional protein expression and reversed mTORC1 dysregulation.
Conclusion:
We provide co-segregation and functional evidence supporting the reclassification of the FLCN p.W376R variant as pathogenic according to ACMG guidelines and report a novel pathogenic mutation, p.Q44*, expanding the known FLCN mutational spectrum. Importantly, FLCN mRNA supplementation restored FLCN expression in vitro, providing preliminary evidence for mRNA-based therapeutic strategies in BHD syndrome.
Introduction
Birt-Hogg-Dubé (BHD) syndrome is a rare autosomal dominant condition caused by germline mutations in the folliculin (FLCN) gene, a tumor suppressor located on the chromosome 17p11.2. 1 The prevalence of BHD in the general population is approximately two cases per million. 2 It is characterized by lung cysts, spontaneous pneumothorax, cutaneous fibrofolliculomas, and renal tumors. Additional manifestations, including parotid oncocytomas, lipoma, thyroid cancer, and colorectal polyps or carcinoma, have been infrequently reported in individuals with BHD, but their inclusion as components of the clinical phenotype of BHD remains unverified.3,4
Over 80% of BHD patients exhibit multiple lung cysts, primarily localized in the lower lobe and near the mediastinum, displaying diverse sizes and shapes.4–6 Cutaneous fibrofolliculomas are regarded as a specific feature of BHD and commonly appear in the head, neck, and upper chest. Individuals diagnosed with BHD syndrome have a significantly elevated seven-fold susceptibility to developing kidney tumors, with the most common pathological manifestations being oncocytic/chromophobe mixed tumors or chromophobe renal cell carcinoma.4,7 The incidence of lung, skin, and renal involvement varies across different regions of the world. Recent studies have found that East Asian cohorts show more pulmonary involvement and fewer skin and kidney manifestations in comparison to Caucasian cohorts.8–11
FLCN is the only known pathogenic gene for BHD syndrome to date, and it is involved in diverse signaling and metabolic pathways and plays an important role in regulating cell growth, proliferation, and survival. Previous studies have demonstrated that FLCN protein impairment leads to hyperactivation of the mechanistic target of rapamycin (mTOR) pathway. Dysregulated mTOR signaling has been closely linked to the pathogenesis of BHD-associated manifestations, including the formation of lung cysts and the development of renal tumors.12–14 According to the Leiden Open Variation Database (http://www.lovd.nl/FLCN), a total of 693 pathogenic FLCN mutations have been identified in the germline of BHD patients, with truncating mutations (frameshift, splice site, nonsense mutations) being the most common and missense mutations being less common (up to March 2026). 15 Relative to truncating mutations, it is difficult to determine the pathogenicity of rare missense mutations without the verification of co-segregation with disease or functional studies.
In vitro-transcribed (IVT) mRNA offers significant promise for treating genetic disorders through supplemental protein expression. Its advantages include direct cytoplasmic protein production without nuclear entry or genomic integration. 16 The therapeutic potential of mRNA was first demonstrated in vivo by Wolff et al., who achieved successful protein expression via direct injection of synthetic mRNA. 17 This foundational work was advanced by Jirikowski et al., who administered vasopressin-encoding mRNA to the mouse hypothalamus, effectively alleviating diabetes insipidus symptoms and providing the first evidence of mRNA-based therapeutic efficacy. 18 Recent studies highlight the successful application of mRNA-based protein replacement in lung diseases, demonstrating efficacy in genetic disorders (cystic fibrosis, surfactant protein B deficiency) and complex respiratory conditions (chronic obstructive pulmonary disease, asthma). 19 Notably, its application for BHD remains unexplored.
Herein, we aim to characterize two distinct Chinese BHD families, with a focus on their clinical features, genetic variants, and functional consequences. Specifically, one family carried the known FLCN p.W376R missense mutation, while the other harbored a novel truncating mutation, p.Q44*. We further sought to confirm the pathogenicity of p.W376R through co-segregation and functional analyses. Finally, we assessed the feasibility of an mRNA-based approach by evaluating FLCN mRNA supplementation in vitro.
Materials and methods
Subjects
This study enrolled two pedigrees with Birt-Hogg-Dubé (BHD) syndrome, designated as Pedigree A and Pedigree B, between February 2023 and December 2023 at the Department of Respiratory and Critical Care Medicine, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology. The proband of Pedigree A was referred to our department in February 2023 due to pneumothorax and multiple cystic lung changes. A three-generation family history was collected from eight relatives, and peripheral blood samples were obtained from seven available individuals. Peripheral blood mononuclear cells (PBMCs) were available only from the proband, as her samples were prospectively collected and processed under RNA-grade conditions; other family members provided blood samples for DNA extraction only and were therefore not included in the RNA analysis. Similarly, the proband of Pedigree B was referred in December 2023 with the same clinical features, after which family histories and peripheral blood samples were obtained from both the proband and her second daughter. The corresponding pedigree charts are illustrated in Figures 1(c) and 2(c), respectively. Peripheral blood samples were obtained in EDTA anticoagulant tubes and maintained at 4°C prior to DNA extraction. Due to the rarity of BHD syndrome, the sample size was determined by the availability of affected individuals from two independent families who consented to participate.
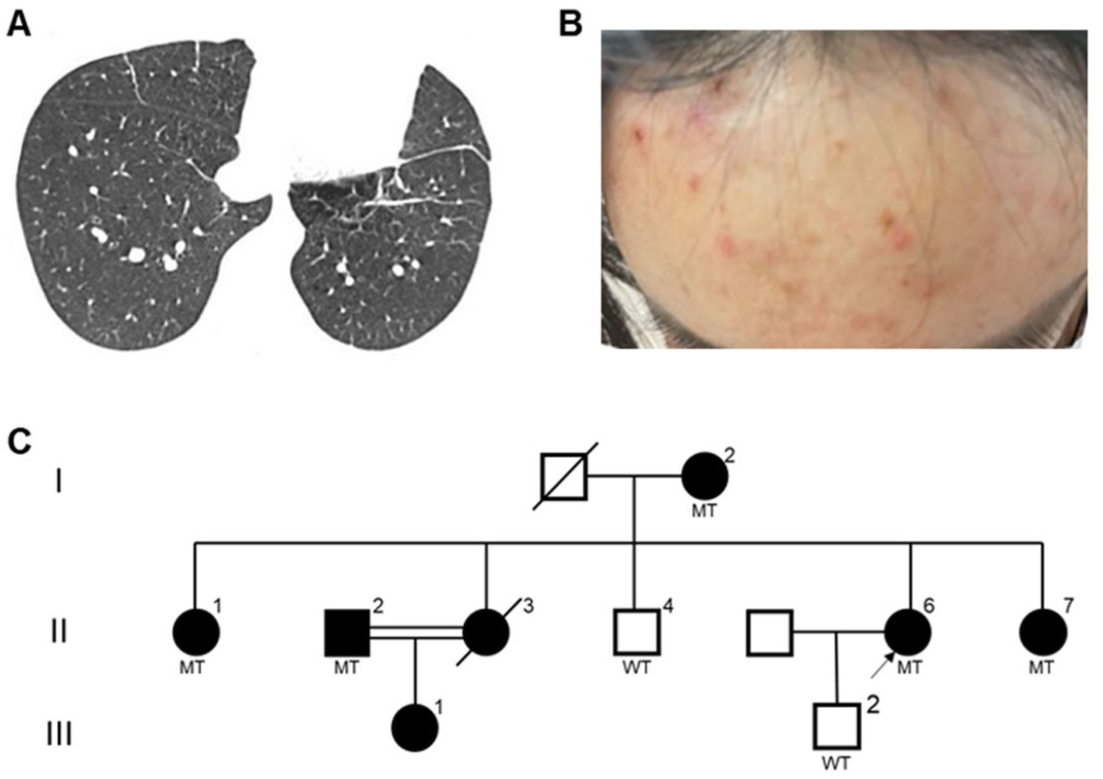
The characteristics of the Pedigree A. (a) Chest CT scan of the proband II-6. (b) Skin lesions of the proband. (c) Family pedigree chart of the proband II-6. Squares and circles indicate male and female, respectively. Arrow indicates the proband. Doubled lines indicate consanguineous marriage. Black-filled symbols represent clinically affected individuals. MT represents mutant, namely heterozygous FLCN p.W376R mutation

The characteristics of the Pedigree B. (a and b) Chest CT scan of (a) the proband I-2 and (b) II-2. (c) Family pedigree chart of the proband I-2. MT represents mutant, namely, heterozygous FLCN p.Q44* mutation.
For functional comparison of FLCN mRNA expression, three healthy control individuals were recruited from the physical examination center of Tongji Hospital. Controls were matched to the proband of Pedigree A by age (±3 years), sex (female), and ethnicity (Han Chinese). All controls had no history of pulmonary cysts, pneumothorax, skin lesions, or renal tumors, and no family history of BHD syndrome or other genetic disorders. Peripheral blood samples were collected for PBMCs isolation and subsequent RNA extraction.
All procedures were performed in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments. All participants signed the written informed consent agreement.
Genetic test
Genomic DNA was extracted from the peripheral blood of the participants using a Qiagen DNA Blood Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s protocol. Whole-exome sequencing (WES) of DNA from the proband was conducted on the Novaseq6000 platform (Illumina, San Diego, USA), 20 with an average sequencing depth of 200×. Several databases, including gnomAD (http://gnomad.broadinstitute.org/), 1000 Genome Project (http://browser.1000genomes.org), OMIM (http://www.omim.org), Clinvar (http://www.ncbi.nlm.nih.gov/clinvar), and HGMD (http://www.hgmd.org) were employed to filter and annotate the mutations. Mutation Taster (http://www.mutationtaster.org/), Poly Phen-2 (http://genetics.bwh.arvard.edu/pph2), 21 SWISS-MODEL (https://swissmodel.expasy.org/), 22 HOPE (https://www3.cmbi.umcn.nl/hope/), I-Mutant2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html), MUPro (https://mupro.proteomics.ics.uci.edu/), 23 and RBPmap (http://rbpmap.technion.ac.il/) 24 were used to predict the impact of variants. The variants were classified to five categories according to the American College of Medical Genetics and Genomics (ACMG) guidelines. 25 Sanger sequencing was performed on an Applied Biosystems 3500 sequencer (Applied Biosystems, USA) to confirm the potentially pathogenic variants identified by WES. 26
Database search for pathogenic missense variants
To investigate the distribution of pathogenic missense variants in FLCN, we systematically queried ClinVar, LOVD database (http://www.lovd.nl/FLCN), and PubMed using the search terms “FLCN” and “missense.” Only variants classified as pathogenic or likely pathogenic were included.
Plasmid construction
The coding sequence of the human FLCN gene was amplified using specific forward (5′-AGTCCAGTGTGGTGGAATTCGCCACCATGAATGCCATCGTGGCTCTCTGC) and reverse (5′-TGGTCTTTGTAGTCCTCGAGGTTCCGAGACTCCGAGGCTGTGGGGCTG) primers. Then the PCR products were purified and cloned into the pCMV-3/FLAG vector to obtain a wild-type plasmid. The W376R and Q44* mutant plasmids were constructed using PCR-based site-directed mutagenesis via the inverse PCR approach. Briefly, the wild-type plasmid was used as the template, and a pair of complementary primers containing the specific mutation was designed for each mutant. The following primers were used: (W376R: forward primer: 5′-GGGAACCAGGTGATCCGGAAAAGCAGAGACGTGGACCTC; reverse primer: 5′-CGTCTCTGCTTTTCCGGATCACCTGGTTCCCCATGAG. Q44*: 5′- GTGAGTAGGCGGAAGAAGAGGAAGGTGGCATT; reverse primer: 5′- TTCTTCCGCCTACTCACCCTGGCCAGGACTGT). PCR was performed using a high-fidelity DNA polymerase (P515, Vazyme, China), followed by digestion of the methylated template plasmid with DpnI (R0176, NEB, USA). The resulting nicked circular DNA containing the desired mutation was transformed into E. coli-competent cells. 27 The recombinant plasmid sequences were verified using Sanger sequencing.
Preparation of FLCN mRNA
The FLCN mRNA generated by in vitro transcription (IVT) is designed to be efficiently translated into functional protein in target cells. It consists of a single-stranded RNA molecule with a 5′ cap and a 3′ poly(A) tail. The coding sequence, or open reading frame (ORF), is flanked by untranslated regions (UTRs) and contains start and stop codons that precisely define the protein sequence. 28 The coding sequence of the human FLCN gene for the in vitro transcription was amplified by PCR using the following primers: 5′-CGAACGATAGCAGCCACCATGAATGCCATCGTGGCT (forward) and 5′-TGAGGCTGGTTGAATTCCCTACTTGTCATCGTCATCCTT (reverse). Then the PCR products were purified and cloned into the E2-W.pUC19-IVT vector (Vazyme, China) located between the 5′-UTR and 3′-UTR and downstream of the T7 promoter via homologous recombination-based seamless cloning using ClonExpress II One Step Cloning Kit (C112, Vazyme, China). 29 DNA templates were obtained through plasmid digestion with HindIII-HF restriction enzyme (R3104, NEB, USA) and then purified using VAHTS DNA Clean Beads (N411-01, Vazyme, China) for IVT. FLCN mRNA was synthesized in vitro using HyperScribe ™ T7 High Yield RNA Synthesis Kit Plus (K1401, APExBIO, USA). The Cap 1 structure was introduced at the 5′ end of the mRNA using the Cap1 analog (B8176, APExBIO, USA), which was added to the IVT reaction. Following transcription, a poly(A) tail was added to the RNA transcripts using the E. coli Poly(A) Polymerase (DD4111, Vazyme, China). Then, the mRNA was purified using the VAHTS RNA Clean Beads (N412, Vazyme, China).
Cell culture
HEK293T cells purchased from the American Type Culture Collection (ATCC, catalog no. CRL-3216, USA) were incubated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented 10% fetal bovine serum (Wisent, Canada) and 1% penicillin/streptomycin at 37°C, 5% CO2 atmosphere.
Plasmid and mRNA transfection
To determine the optimal transfection dose, HEK293T cells were transfected with increasing amounts of FLCN mRNA (1.25, 2.5, 3.75, and 5 μg) using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer’s instructions. A dose of 3.75 μg was selected as the optimal dose for subsequent experiments. For functional experiments, cells were plated in 12-well plates overnight and transfected with indicated plasmids using Lipofectamine 3000 transfection reagent based on the manufacturer’s protocol. Following 24-h incubation, cells were transfected with FLCN mRNA (3.75 μg). After a further 48-hour culture period, cells were harvested for downstream analysis. The same reagent kit and identical procedures were used to prepare FLCN mRNA for each experiment. All transfection experiments were independently performed three times.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol Reagent (Takara, Dalian, China). The complementary DNA (cDNA) was synthesized by PrimeScript RT reagent Kit (Vazyme, Nanjing, China). qRT-PCR was performed on the CFX Connect Real-Time System (Bio-Rad, USA) utilizing SYBR qPCR Master Mix (Vazyme, Nanjing, China). Primer sequences for qRT-PCR are as follows: FLCN forward primer, TCTTCAGCATTGTCCGCCAG, reverse primer, AGTTGATGAGGTAGATCCGGTC, ACTB forward primer, AGAAAATCTGGCACCACACCT, reverse primer, GATAGCACAGCCTGGATAGCA. The relative mRNA expression was normalized to ACTB and calculated using the 2−ΔΔCT method. 30 For human PBMC samples, each sample was tested in triplicate technical replicates; for in vitro transfection experiments, each group had duplicate technical replicates, and the experiment was independently repeated three times.
Western blot analysis
Total protein was extracted from the harvested HEK293T cells using RIPA lysis buffer (Aspenbio, Wuhan, China), then the protein was separated in 12% SDS-PAGE and transferred to PVDF membranes. Following blocking with 5% non-fat milk at room temperature for 1 h, the membranes were incubated with primary antibodies (anti-FLAG mAb, Proteintech, 1:4000; anti-mTOR pAb, Proteintech, 1:5000; anti-Phospho-mTOR pAb, Proteintech, 1:10,000; anti-GAPDH mAb, Proteintech, 1:4000), respectively, overnight at 4°C. Subsequently, the membranes were washed with TBS-Tween three times and exposed to horseradish peroxidase-conjugated goat anti-mouse secondary antibody (Aspenbio, 1:4000) for 1 h. Protein bands were detected using ECL reagent (MCE, USA).
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 9.0. Student’s t-test was used to compare the significance between groups, and one-way ANOVA was used for comparisons among multiple groups. Data were expressed as mean ± SEM, p value < 0.05 was considered statistically significant.
Results
Clinical characteristics
Pedigree A: The proband II-6, a 33-year-old female, underwent surgical bullectomy for pneumothorax and multiple pulmonary bullae. Postoperative chest computed tomography (CT) revealed bilateral cysts predominantly distributed in the basal medial lung regions (Figure 1(a)). She was subsequently referred to the Department of Respiratory and Critical Care Medicine for further diagnostic evaluation and management. The patient reported a 4-month history of exertional dyspnea without obvious precipitating factors. She denied any history of smoking, alcohol consumption, or other significant medical conditions. General physical examination indicated multiple red papules on her face and arms, which were diagnosed as folliculitis by a professional dermatologist (Figure 1(b)). Importantly, her mother, sisters, cousin, and niece also suffered from lung lesions (pulmonary cysts and/or spontaneous pneumothorax). Based on the family history and clinical and radiological manifestations, we suspected a family hereditary disease, particularly BHD. The clinical data of affected individuals in Pedigree A are shown in Table 1. All seven patients had bilateral pulmonary cysts or experienced pneumothorax, with four having had multiple episodes of pneumothorax (one of whom passed away due to pneumothorax) and two having had a single pneumothorax. I-2, II-1, and II-2 received conservative management (thoracic drainage or observation) for pneumothorax, whereas II-6 and III-1 underwent thoracoscopic bullectomy as an intervention for their first occurrence of pneumothorax. The proband suffered from folliculitis rather than typical fibrofolliculomas or trichodiscomas of BHD; the other affected individuals did not have skin involvement. No significant renal abnormality was observed in II-2, II-5, and II-6; unfortunately, the remaining individuals did not consent to renal ultrasound examination.
Clinical data of affected individuals.

Deceased due to pneumothorax.
F, female; M, male; NA, not available.
Pedigree B: Three months prior to presentation, the proband I-2 had received closed thoracic drainage for a left-sided pneumothorax. Chest CT at that time revealed a left-sided pneumothorax and bilateral multiple pulmonary bullae (Figure 2(a)). The patient presented to our hospital to determine the underlying cause. She had no history of smoking, alcohol consumption, or other chronic diseases. Physical examination was unremarkable. Notably, her second daughter also had pulmonary cysts (Figure 2(b)) but no history of spontaneous pneumothorax. Neither individual exhibited cutaneous fibrofolliculomas or renal involvement. Table 1 displays the clinical manifestations of affected members in this family.
Genetics analysis
Pedigree A: WES of the proband II-6 identified a heterozygous FLCN variant (c.1126T>C, p.W376R) in exon 10, which was validated by Sanger sequencing. No additional potential pathogenic variants or copy number variations (CNVs) were identified. Notably, this missense variant was absent from population databases (gnomAD and 1000 Genomes Project), indicating no reported allele frequency in control populations. To evaluate the potential impact of this variant, multiple computational analyses were performed. First, Mutation Taster and PolyPhen-2 predicted that the variant was pathogenic and highly conserved across multiple species (Figure 3(a)). Using the RBPmap tool, we analyzed the wild-type and mutant FLCN sequences (transcript NM_144997.7) within a 180‑bp window centered on the variant. The analysis revealed that the c.1126T>C (p.W376R) mutation introduces a binding motif for the RNA-binding protein RBM6 (Z‑score = 2.042, p < 0.05), which was absent in the wild-type sequence. The acquisition of an RBM6-binding site could potentially influence mRNA stability or translational efficiency. Subsequently, we employed SWISS-MODEL and HOPE to construct wild-type and mutant protein models and to predict the impact of this amino acid substitution on protein structure, respectively (Figure 3(b)). The charge, size, and hydrophobicity differed between wild-type and mutant residue, which could disturb the cDENN FLCN/SMCR8-type (annotated in UniProt) domain and abolish its function. Further stability assessments utilizing I-Mutant 2.0 and MUpro platforms suggested that this amino acid substitution reduces protein stability. It was classified as a variant of uncertain significance (VUS): PM2+PP3. Next, we conducted co-segregation analysis to evaluate its pathogenicity. As shown in Figure 1(c), a total of seven individuals of this family consented to validate this variant; patients I-2, II-1, II-2, II-6, and II-7 carried the p.W376R mutation, whereas unaffected members II-4 and III-2 did not carry this mutation. Thus, these co-segregation data can be regarded as stronger evidence.
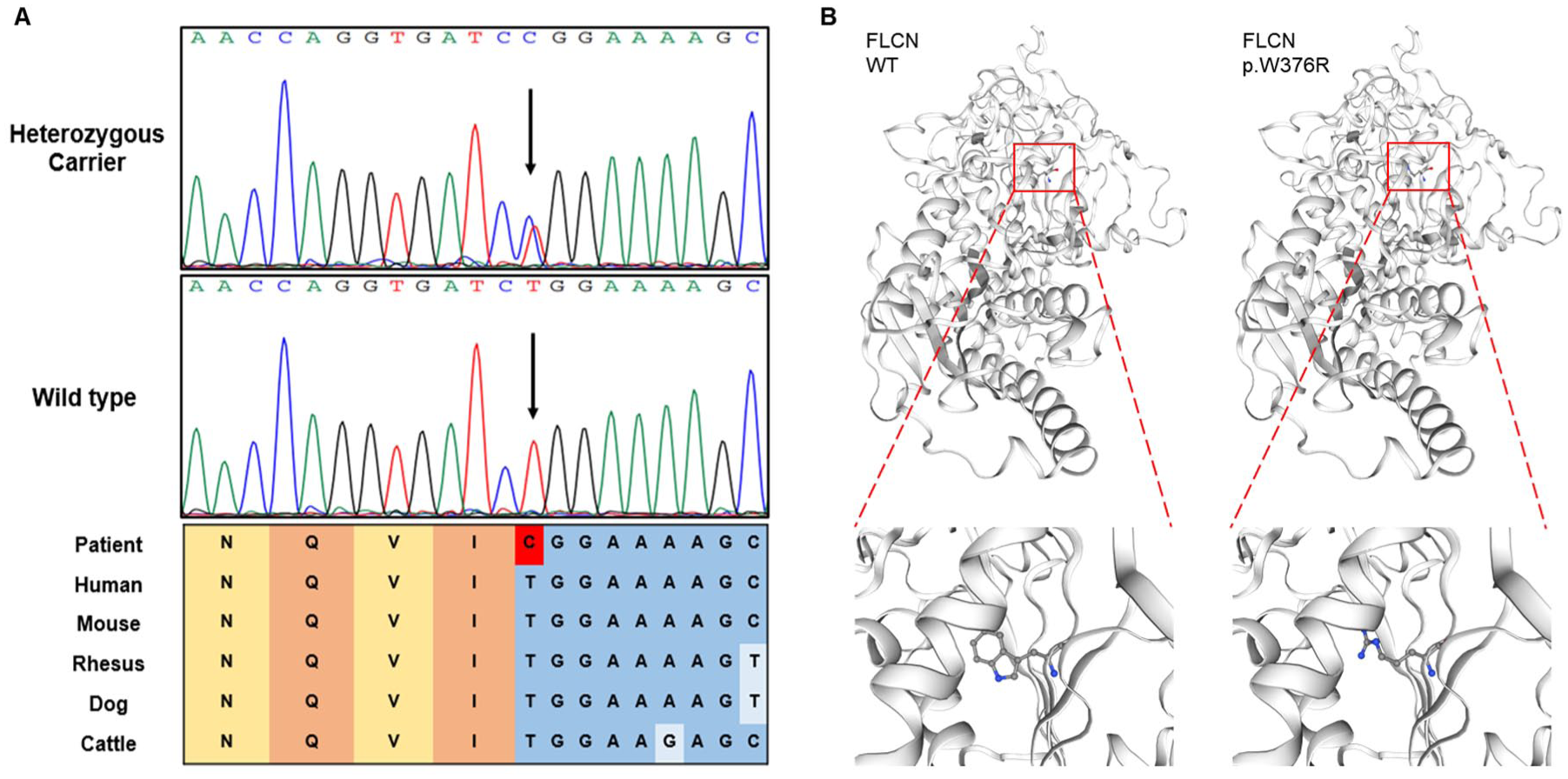
Sanger sequencing and prediction analyses. (a) Sanger sequencing results of the family. Arrows indicate the position of the mutation. Colored blocks indicate evolutionary conservation around the mutation among multiple species. (b) Three-dimensional protein structure prediction of wild-type (left) and mutant proteins (right).
Pedigree B: WES performed on peripheral blood from the proband (I-2) identified a heterozygous nonsense mutation, c.130C>T (p.Q44*), within exon 4 of the FLCN coding region. No further potentially pathogenic variants or CNVs were detected. This mutation introduces a premature termination codon at codon 44, resulting in a truncated protein product. In addition, the variant was not found in general population databases such as gnomAD and 1000 Genomes Project. Based on ACMG criteria, this mutation is classified as likely pathogenic (PVS1+PM2). To our knowledge, the FLCN p.Q44* variant is reported for the first time. Furthermore, Sanger sequencing confirmed that individual II-2 also carried the p.Q44* mutation (Figure 2(c)). As other individuals in this pedigree declined genetic testing, co-segregation analysis between the genotype and phenotype could not be performed.
Functional validation of FLCN p.W376R pathogenicity
To further explore whether the p.W376R mutation affects the expression levels of FLCN, we first performed qRT-PCR to measure FLCN mRNA levels in PBMCs from the proband (Pedigree A) and three matched healthy individuals. The results revealed that the mRNA expression of FLCN in the proband was lower than in healthy controls (Figure 4(a)).

Functional analysis of FLCN mutations and mRNA therapeutic intervention. (a) Quantitative PCR analysis comparing FLCN mRNA expression in peripheral blood PBMCs between healthy controls and the proband II-6. (b) qPCR analysis of FLCN mRNA expression in HEK293T cells transfected with empty vector, wild-type FLCN plasmid, or FLCN p.W376R plasmid. (c) Representative immunoblots and quantitative analysis of FLCN protein expression in HEK293T cells transfected with varying doses of FLCN mRNA. (d) Representative immunoblots and quantitative analysis of FLCN, mTOR, and p-mTOR expression in HEK293T cells transfected with either an empty vector, wild-type plasmid, p.W376R plasmid, or p.Q44* plasmid, followed by treatment with or without FLCN mRNA therapy. Data are presented as mean ± SEM from three independent experiments. ****p < 0.0001 versus the wild-type group.
HEK293T cells were transfected with the pCMV-3/FLAG empty plasmid, the pCMV-3/FLAG-FLCN wild-type plasmid, or the pCMV-3/FLAG-FLCN W376R plasmid. Quantitative RT-PCR analysis demonstrated significantly attenuated FLCN mRNA expression in p.W376R-transfected cells compared to the wild-type group (Figure 4(b)). Consistent with transcriptional downregulation, Western blot showed a distinct FLCN protein band in HEK293T cells transfected with wild-type FLCN, whereas the expression levels of FLCN protein in the p.W376R variant group were almost undetectable (Figure 4(d)). Collectively, these functional data provide definitive evidence supporting pathogenicity classification of the p.W376R variant under ACMG/AMP PS3 criterion (functional evidence of deleterious impact).
FLCN mRNA intervention rescues pathogenic variant-induced protein deficiency and mTOR dysregulation
To rescue the mutation-induced downregulation of FLCN expression, we synthesized FLCN mRNA by in vitro transcription. Subsequently, we examined whether the FLCN mRNA could produce functional FLCN protein. We transfected HEK293T cells with varying amounts of FLCN mRNA. Western blot analysis revealed that the FLCN protein level was significantly higher than that in the control group, and the highest protein expression level was observed following transfection of 3.75 μg of FLCN mRNA (Figure 4(c)). Based on these results, 3.75 μg was selected as the standard transfection dose for all subsequent experiments.
To further investigate whether FLCN mRNA could rescue the protein deficiency caused by the p.W376R and p.Q44* variants, HEK293T cells were transfected with the pCMV-3/FLAG empty plasmid, the pCMV-3/FLAG-FLCN wild-type plasmid, the pCMV-3/FLAG-FLCN W376R plasmid, or the pCMV-3/FLAG-FLCN Q44* plasmid. Following 24-h incubation, cells were transfected with or without FLCN mRNA. Quantitative Western blot analysis demonstrated significant restoration of FLCN protein expression in both p.W376R and p.Q44* variant groups, reaching levels comparable to the wild-type FLCN group. Furthermore, we assessed mTOR and phosphorylated mTOR (p-mTOR) levels across all conditions. Both mutant groups showed a trend toward hyperactivation of mTOR signaling, as indicated by an increased p‑mTOR/mTOR ratio. Critically, therapeutic intervention with FLCN mRNA partially suppressed this aberrant p-mTOR elevation, with a trend toward reduction in the p.W376R group (p = 0.0957) and a statistically significant reduction in the p.Q44* group (p = 0.0202) (Figure 4(d)).
Discussion
Our study reported a BHD family harboring the FLCN c.1126T>C (p.W376R) mutation, previously categorized as “VUS” (PM2+PP3). Of note, the FLCN c.1126T>A (p.W376R) variant has been submitted to ClinVar as pathogenic by Juntendo University (RCV003326278). However, that submission did not provide detailed family history, co‑segregation, or functional validation data, nor did it specify the criteria used for the assertion. Using supplementary co-segregation evidence and functional studies, we demonstrated that this variant also satisfies the PS3 and PP1_strong criteria outlined in the ACMG guidelines; therefore, we recommended reclassifying this variant as “pathogenic” (PS3+PP1_strong+PM2+PP3). In addition, a novel FLCN p.Q44* truncating variant was identified in another pedigree. Critically, mRNA supplementation effectively rescued FLCN protein expression in vitro for both variants.
BHD syndrome is a heterogeneous disease characterized by varying degrees of lung, skin, and/or renal involvement.1,3 It is not easy to differentiate BHD from other cystic lung diseases (lymphangioleiomyomatosis, Langerhans cell histiocytosis, and so on) in patients who present only with bilateral lung cysts in the absence of skin lesions or renal tumors. In this study, all affected individuals in the two families presented with pulmonary manifestations; neither typical BHD skin lesions nor renal cancer were observed. These manifestations were consistent with previous studies in the Chinese cohort.8,31,32 Therefore, genetic screening is an effective method for early diagnosis of BHD syndrome, particularly in those patients with a positive family history but no characteristic skin or renal lesions.
The c.1285dupC/delC in exon 11 is the most frequent mutation among BHD patients in China and other nations. However, variations in the FLCN mutation spectrum also exist. For example, a significant proportion of the mutations found in Chinese patients have not been documented in other ethnic groups, whereas the two mutation hotspots (c.1533_1536delGATG and c.1347_1353dupCCACCCT) reported in Japanese patients were not detected in Chinese patients. 8 In the present study, we discovered a novel truncating mutation, FLCN c.130C>T (p.Q44*), in Pedigree B, which expands the mutational spectrum associated with BHD. In addition, we identified a variant in Pedigree A: FLCN c.1126T>C (p.W376R), which has not been previously reported in Chinese patients. More than half of the patients harboring this mutation displayed multiple episodes of pneumothorax, which contrasts with the observations of Sattler et al., who described this mutation in three BHD patients presenting with a single pneumothorax. 33 Since the specific clinical data pertaining to patients, including skin manifestations, renal involvement, age, gender, and other relevant factors, were not provided in Sattler’s research, we were unable to compare similarities and differences between the two studies. Notably, there are eight affected individuals (five in this pedigree and three from another family) belonging to two different races, supporting that co-segregation with disease can be used as strong evidence toward pathogenicity. We further analyzed the distribution of known pathogenic missense variants in FLCN. A systematic review of ClinVar, LOVD, and PubMed identified a total of 29 pathogenic/likely pathogenic missense variants (Table S1). Among these, the majority were located in exons 4 and 6, whereas exon 10 contained only a few pathogenic missense variants.
Some studies have suggested that truncating mutations of FLCN, as described in most patients, can result in functional deficiency of the gene product, while missense mutations may impair protein stability, thereby leading to BHD phenotypes.34,35 Consistent with this paradigm, the p.Q44* variant introduces a premature termination codon at residue 44, resulting in truncated protein synthesis and predicted loss of functional FLCN. However, the impact of the p.W376R variant on the transcription and translation processes of the FLCN protein remains uncertain. In this study, we performed functional experiments in vitro to examine the impact of this missense mutation on gene expression. The results revealed that p.W376R could diminish the mRNA and protein expression of FLCN, indicating that it impairs the stability of FLCN mRNA and protein.
Given that FLCN is the only known causative gene for BHD syndrome, 1 therapeutic restoration of FLCN function represents a promising intervention strategy. mRNA-based protein replacement represents a promising therapeutic strategy for BHD syndrome due to its advantages of no risk of gene integration, transient yet regulatable protein expression, and rapid metabolic clearance. 28 Despite these mechanistic advantages, mRNA therapeutic approaches remain unexplored for this disorder. Herein, we implemented in vitro-transcribed FLCN mRNA delivery systems to rescue disease-associated variants (p.W376R and p.Q44*). Experimental data indicated that the delivered mRNA effectively generated functional folliculin protein and partially corrected dysregulated mTOR signaling. These findings provide initial evidence supporting further preclinical evaluation of mRNA supplementation in disease-relevant models.
There are some limitations in this research. First, within the two pedigrees analyzed, clinical data from certain family members were unavailable due to their refusal to participate. The current sample size is insufficient to establish the relationship between p.W376R/p.Q44* and phenotypic features, necessitating future investigation with larger cohorts. Second, all functional experiments were performed in HEK293T cells. Although this model is suitable for initial validation, it does not fully reflect the physiological context of BHD. Further validation in disease-relevant cell types and preclinical animal models is needed. In addition, validation of the therapeutic efficacy of synthetic FLCN mRNA was restricted to the p.W376R and p.Q44* variants and to in vitro models only. The general applicability of mRNA-based intervention for BHD requires further validation across a broader spectrum of mutations and in preclinical models.
Conclusion
In summary, we identified a pathogenic FLCN variant, p.W376R, in a Chinese BHD family, providing the first co-segregation and functional data for this mutation, and discovered a novel FLCN mutation, p.Q44*, in another pedigree. These findings contribute to the expanding spectrum of pathogenic mutations associated with FLCN and offer valuable insights for genetic counseling. Furthermore, we present the first experimental evidence demonstrating that FLCN mRNA intervention can restore FLCN function in vitro, offering preliminary evidence for exploring mRNA-based strategies in BHD.
Supplemental Material
sj-docx-1-tar-10.1177_17534666261451630 – Supplemental material for Novel FLCN mutations in Birt-Hogg-Dubé patients and potential intervention of FLCN mRNA
Supplemental material, sj-docx-1-tar-10.1177_17534666261451630 for Novel FLCN mutations in Birt-Hogg-Dubé patients and potential intervention of FLCN mRNA by Wenxue Bai, Mengyao Guo, Lijuan Hua, Xuezhao Wang, Lirong Chen, Bingyi Liu, Yi Wang, Ying Zhou, Qi Wang, Ni Zhang, Lan Chen, Min Wu, Zongzhe Li and Min Xie in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-2-tar-10.1177_17534666261451630 – Supplemental material for Novel FLCN mutations in Birt-Hogg-Dubé patients and potential intervention of FLCN mRNA
Supplemental material, sj-docx-2-tar-10.1177_17534666261451630 for Novel FLCN mutations in Birt-Hogg-Dubé patients and potential intervention of FLCN mRNA by Wenxue Bai, Mengyao Guo, Lijuan Hua, Xuezhao Wang, Lirong Chen, Bingyi Liu, Yi Wang, Ying Zhou, Qi Wang, Ni Zhang, Lan Chen, Min Wu, Zongzhe Li and Min Xie in Therapeutic Advances in Respiratory Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.