
Abstract
Background:
Interstitial pneumonia with autoimmune features (IPAF) represents a heterogeneous entity overlapping with connective tissue disease-associated interstitial lung disease (CTD-ILD); however, its clinical course and prognostic determinants remain incompletely characterized.
Objective:
This study aimed to compare baseline characteristics, outcomes, and predictors of poor prognosis between IPAF and CTD-ILD, and to evaluate the prognostic performance of selected inflammatory indices.
Design:
This study was designed as a single-center, retrospective cohort analysis evaluating clinical characteristics, radiological features, and prognostic outcomes in patients with interstitial lung disease.
Methods:
This retrospective cohort study included 88 patients with ILD, categorized as IPAF (n = 30) or CTD-ILD (n = 58), including systemic sclerosis, rheumatoid arthritis, and Sjögren’s disease-associated ILD. Baseline demographic, clinical, radiological, and laboratory data were systematically analyzed. Poor prognosis was defined as the occurrence of at least one of the following during follow-up: >10% decline in forced vital capacity (FVC), development of a radiological progression, or the need for treatment escalation. Univariable and multivariable logistic regression analyses were performed to identify predictors of poor prognosis. Discriminative performance of the Gender–Age–Physiology (GAP) score, Lung Immune Prognostic Index (LIPI), and derived neutrophil-to-lymphocyte ratio (dNLR) was evaluated using receiver operating characteristic (ROC) curve analysis.
Results:
Patients with IPAF were older at diagnosis compared with CTD-ILD (median 68.5 vs 56.0 years, p < 0.001), while female predominance was higher in CTD-ILD (81.0% vs 56.7%, p = 0.029). The GAP score was higher in IPAF, whereas anti-CCP positivity was more common in CTD-ILD. Despite these differences, clinical outcomes were comparable between groups, with no significant differences in FVC decline, development of radiological progression, treatment escalation, antifibrotic initiation, or composite poor prognosis. In univariable analysis, an increased persistent erythrocyte sedimentation rate (ESR) and C-reactive protein were associated with poor prognosis. In multivariable analysis, persistent ESR elevation remained an independent predictor (OR 3.25, 95% CI 1.15–9.23, p = 0.027). ROC analysis showed limited discriminative performance (AUC < 0.60) for GAP, LIPI, and dNLR.
Conclusion:
Although IPAF and CTD-ILD differ in baseline phenotype, their clinical trajectories appear similar. Persistent systemic inflammation, reflected by sustained ESR elevation, independently predicts adverse outcomes, while commonly used prognostic scores demonstrate limited utility.
Keywords
Introduction
The pathogenesis of connective tissue disease-associated interstitial lung disease (CTD-ILD) is considered to be primarily driven by immune-mediated inflammation rather than intrinsic fibrotic remodeling, in contrast to the pattern typically seen in idiopathic pulmonary fibrosis (IPF). Nevertheless, a subset of patients with idiopathic interstitial pneumonia (IIP) exhibit clinical or serological features suggestive of CTD—such as inflammatory arthritis or characteristic cutaneous manifestations—yet do not fulfill the established classification criteria for a defined CTD. This clinical entity has been described as interstitial pneumonia with autoimmune features (IPAF). 1 Patients with IPAF frequently exhibit autoantibody profiles similar to those observed in CTD-ILD. The other shared feature of these entities is the presence of a nonspecific interstitial pneumonia (NSIP) pattern on lung imaging, which has been observed more commonly among patients with CTD-ILD and IPAF compared with idiopathic cases. 2 Moreover, improvements in lung function following immunosuppressive therapy have been reported in this population. Together, these findings underscore the need for careful clinical monitoring and suggest that early immunosuppressive treatment may be beneficial, even in the absence of overt extrapulmonary autoimmune features. 3 The clinical management of IPAF is inherently complex and requires close coordination across specialties, particularly pulmonology, rheumatology, and radiology. Within this integrated, multidisciplinary setting, continuous and careful monitoring of disease behavior is essential, as it underpins risk stratification and informs individualized clinical decision-making. 4
Contrary, the literature presents heterogeneous findings regarding prognostic indicators in this field. In a large cohort, compared with patients with CTD-ILD, those with IPAF were older and more frequently male. Mortality was higher in IPAF than in CTD-ILD. 5 In another study, which included 72 patients with IPAF, the use of glucocorticoids at doses exceeding 20 mg/day was associated with increased mortality. 6 In another study, advanced age and a history of smoking were also identified as predictors of poorer prognosis. 7 Nevertheless, IPAF is generally considered to be associated with a less severe pulmonary disease course compared with IPF. 8
In the present study, our objective was to compare baseline clinical scores, serological markers, and subsequent disease-related outcomes—including functional decline of FVC, radiological progression, and a need for treatment escalation among patients with CTD-ILD and IPAF. Furthermore, we sought to determine whether specific clinical scores or biomarkers could help enable the early identification of patients at increased risk of an unfavorable prognosis.
Methods
Study design and patient selection criteria
This single-center retrospective cohort study was conducted in the Rheumatology Department of Pamukkale University Hospital. All participants had been followed at our center over the past 5-year period, between January 2021 and January 2026. Based on detailed clinical, serological, and radiological assessments, patients were categorized as either IPAF or CTD-ILD. Patients were considered eligible if they were aged 18 years or older, had ILD confirmed by high-resolution computed tomography (HRCT), and fulfilled the classification criteria for either IPAF, 9 or a defined CTD-ILD. 10 In addition, inclusion required the availability of baseline demographic, clinical, and serological data at the time of diagnosis, as well as documented longitudinal follow-up in the rheumatology clinic. Patients were excluded if ILD was attributed to non-autoimmune causes, such as infection, malignancy, drug-induced pulmonary toxicity, or IPF. In addition, individuals with incomplete baseline clinical or laboratory data, or insufficient longitudinal follow-up information in the medical records, were not included in the analysis. As this was a retrospective exploratory study, no formal a priori sample size calculation was undertaken. All consecutive patients who met the predefined inclusion and exclusion criteria during the study period were enrolled, ensuring an unselected and representative cohort. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement (Figure 1; Supplemental Material). 11
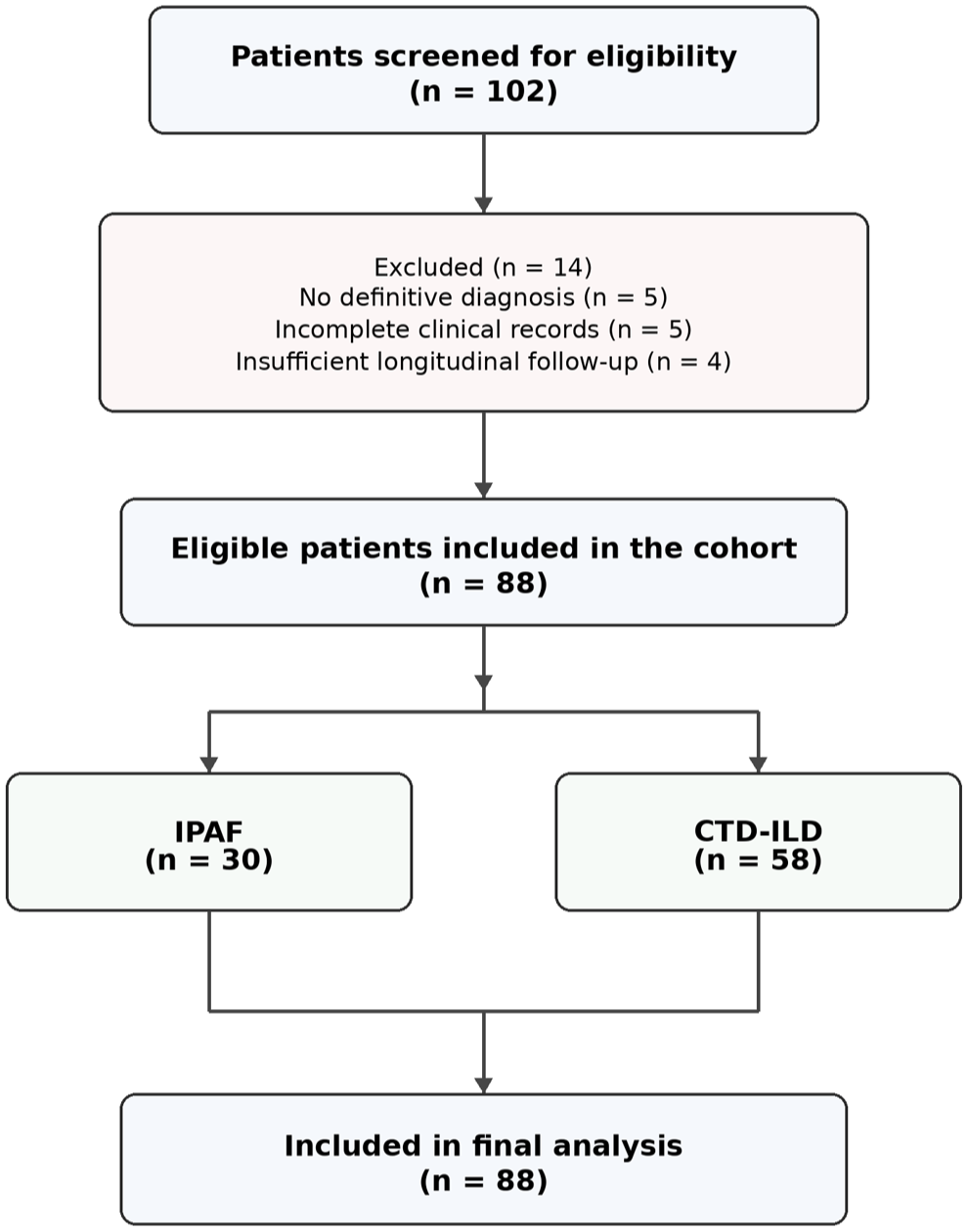
Study flow diagram illustrating patient screening, exclusion, cohort allocation, and final analysis. Of 102 patients with ILD screened for eligibility, 14 were excluded because of the absence of a definitive diagnosis (n = 5), incomplete clinical records (n = 5), or insufficient longitudinal follow-up (n = 4). The final cohort consisted of 88 patients, including 30 patients with IPAF and 58 patients with CTD-ILD, all of whom were included in the final analyses.
Baseline assessment of patient data and prognostic scores
Baseline variables corresponded to data recorded at the time of ILD diagnosis. Demographic characteristics, clinical findings, smoking status, comorbidities, and autoantibody profiles were extracted from medical records. Radiologic patterns were categorized based on HRCT findings. Baseline pulmonary function tests included FVC and diffusing capacity of the lung for carbon monoxide (DLCO) expressed as percent predicted. Prognostic indices and composite scores were calculated using available variables.
The Gender–Age–Physiology (GAP) index was determined for all patients to assess disease severity and estimate mortality risk in ILD. This score incorporates gender, age, FVC % predicted, and DLCO % predicted. Based on the total score, patients were classified into GAP stages I–III, corresponding to progressively higher mortality risk, as originally described by Ley et al. 12
The Lung Immune Prognostic Index (LIPI) was further evaluated as an indicator of systemic inflammatory status. This index is based on two components: the derived neutrophil-to-lymphocyte ratio (dNLR) and serum lactate dehydrogenase (LDH) levels. The dNLR was obtained by dividing the absolute neutrophil count by the difference between the total white blood cell count and the neutrophil count. In accordance with the original LIPI classification, patients were categorized into three prognostic groups: good (score 0), intermediate (score 1), and poor (score 2). A score of one point was assigned for a dNLR >3 and one point for LDH levels exceeding the upper limit of normal, resulting in a cumulative score ranging from 0 to 2. 13
Persistent erythrocyte sedimentation rate (ESR) and c-reactive protein (CRP) elevation were defined as values above the laboratory upper limit of normal on at least two consecutive visits separated by at least 3 months during follow-up.
Parameters at the follow-up and definition of outcomes
Pulmonary function decline was defined as a relative reduction of ⩾10% in FVC compared with baseline values, assessed through serial measurements. Functional deterioration, however, was primarily anchored to a ⩾10% relative decline in FVC, given its greater consistency and clinical interpretability across longitudinal follow-up compared to DLCO. This approach was necessary because a subset of patients was unable to tolerate or reliably perform the DLCO test, precluding consistent longitudinal assessment in all cases.
Radiological progression was defined as an increased extent of fibrotic abnormalities on HRCT, including reticulation, traction bronchiectasis, or honeycombing, and/or newly developed or increased ground-glass opacities considered consistent with disease progression after multidisciplinary review.
Treatment escalation was defined as intensification of therapy due to clinically relevant disease worsening, including initiation of antifibrotic therapy, dose escalation of immunosuppressive treatment (e.g., mycophenolate mofetil up to 3 g/day), or escalation/addition of rituximab (e.g., 2 × 1000 mg). Antifibrotic therapy was initiated in patients with a progressive fibrotic phenotype, based on radiological progression of fibrotic abnormalities and/or functional decline. Escalation of anti-inflammatory or immunosuppressive therapy was considered when disease worsening was interpreted as predominantly inflammatory, including increased disease-related ground-glass abnormalities, progressive respiratory symptoms, or functional deterioration. Inflammatory markers, including ESR and CRP, were considered supportive laboratory parameters but were not used as standalone criteria for treatment escalation.
Patients were followed in the rheumatology clinic, and the duration of follow-up was documented for each individual. The composite poor-prognosis endpoint was designed to capture clinically meaningful deterioration in real-world practice, recognizing that progression in autoimmune-related ILD may occur through multiple pathways. This endpoint was considered present if at least one of the following occurred during follow-up: a relative decline exceeding 10% in FVC, radiological evidence of progression on HRCT, or the need for treatment escalation, defined as initiation of antifibrotic therapy or intensification of immunosuppressive treatment.
Statistical analysis
All analyses were performed using SPSS (IBM Corp., Armonk, NY, USA). Continuous variables were summarized as medians with interquartile ranges (IQRs), whereas categorical variables were reported as frequencies and percentages. Comparisons between groups were conducted using the Mann–Whitney U test for continuous variables that were not normally distributed and the chi-square test for categorical variables; Fisher’s exact test was used when necessary. To investigate factors associated with poor prognosis, univariable logistic regression analyses were initially performed, followed by multivariable models incorporating clinically relevant variables to determine independent predictors. Variables for multivariable analysis were selected according to clinical relevance and findings from univariable analyses. Given the limited number of outcome events, the multivariable model was deliberately restricted to a small number of clinically relevant covariates to minimize overfitting. Closely related variables were not entered simultaneously in order to reduce collinearity. The discriminative ability of the models was assessed using ROC analysis. Findings were presented as odds ratios (ORs) with corresponding 95% confidence intervals (CIs), and a two-tailed p-value <0.05 was considered statistically significant. All statistical analyses were carried out using appropriate software.
Missing baseline variables were limited and primarily affected DLCO measurements, which were available for 25/30 IPAF patients and 54/58 CTD-ILD patients. Analyses were performed using complete-case data, and no imputation procedures were applied.
Results
The whole study population involved; patients with IPAF (n = 30) and those with CTD-ILD, including systemic sclerosis (SSc), rheumatoid arthritis (RA), and Sjögren’s disease (SjD) (n = 58). As presented in Table 1, patients with IPAF were significantly older at diagnosis and exhibited higher GAP scores compared with the CTD-ILD group, suggesting that these patients may present with a less favorable baseline respiratory risk profile. In contrast, the proportion of female patients was significantly higher among individuals with CTD-ILD. Notably, the median follow-up duration was shorter in the IPAF group, as shown in Table 1.
Baseline demographic, serological, radiological, pulmonary function, and inflammatory characteristics of patients with interstitial pneumonia with autoimmune features IPAF and CTD-ILD.
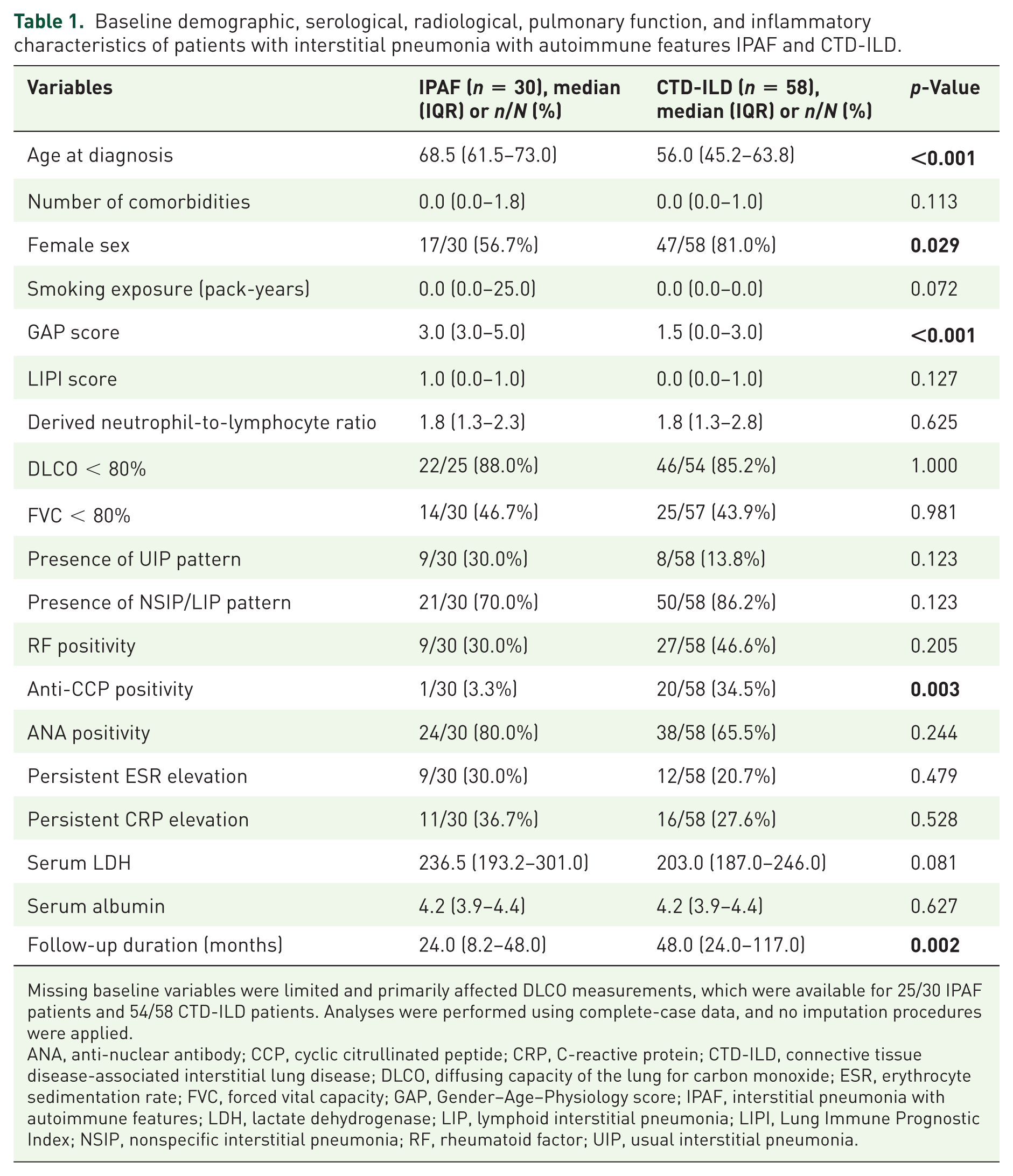
Missing baseline variables were limited and primarily affected DLCO measurements, which were available for 25/30 IPAF patients and 54/58 CTD-ILD patients. Analyses were performed using complete-case data, and no imputation procedures were applied.
ANA, anti-nuclear antibody; CCP, cyclic citrullinated peptide; CRP, C-reactive protein; CTD-ILD, connective tissue disease-associated interstitial lung disease; DLCO, diffusing capacity of the lung for carbon monoxide; ESR, erythrocyte sedimentation rate; FVC, forced vital capacity; GAP, Gender–Age–Physiology score; IPAF, interstitial pneumonia with autoimmune features; LDH, lactate dehydrogenase; LIP, lymphoid interstitial pneumonia; LIPI, Lung Immune Prognostic Index; NSIP, nonspecific interstitial pneumonia; RF, rheumatoid factor; UIP, usual interstitial pneumonia.
In the initial treatment phase, mycophenolate mofetil (MMF) was the most frequently preferred therapy in the IPAF group, whereas cyclophosphamide and rituximab were used more commonly in the CTD-ILD group. During subsequent lines of therapy, MMF continued to be the predominant treatment in patients with IPAF, while rituximab appeared to be used more frequently among patients with CTD-ILD. Throughout the follow-up period, no statistically significant differences were identified between the two groups with regard to decline in FVC, development of radiological progression, need for treatment escalation, or requirement for antifibrotic therapy. Similarly, the composite poor prognosis endpoint did not reach statistical significance, as shown in Table 2.
Longitudinal disease outcomes and progression-related parameters in the IPAF and CTD-ILD groups.
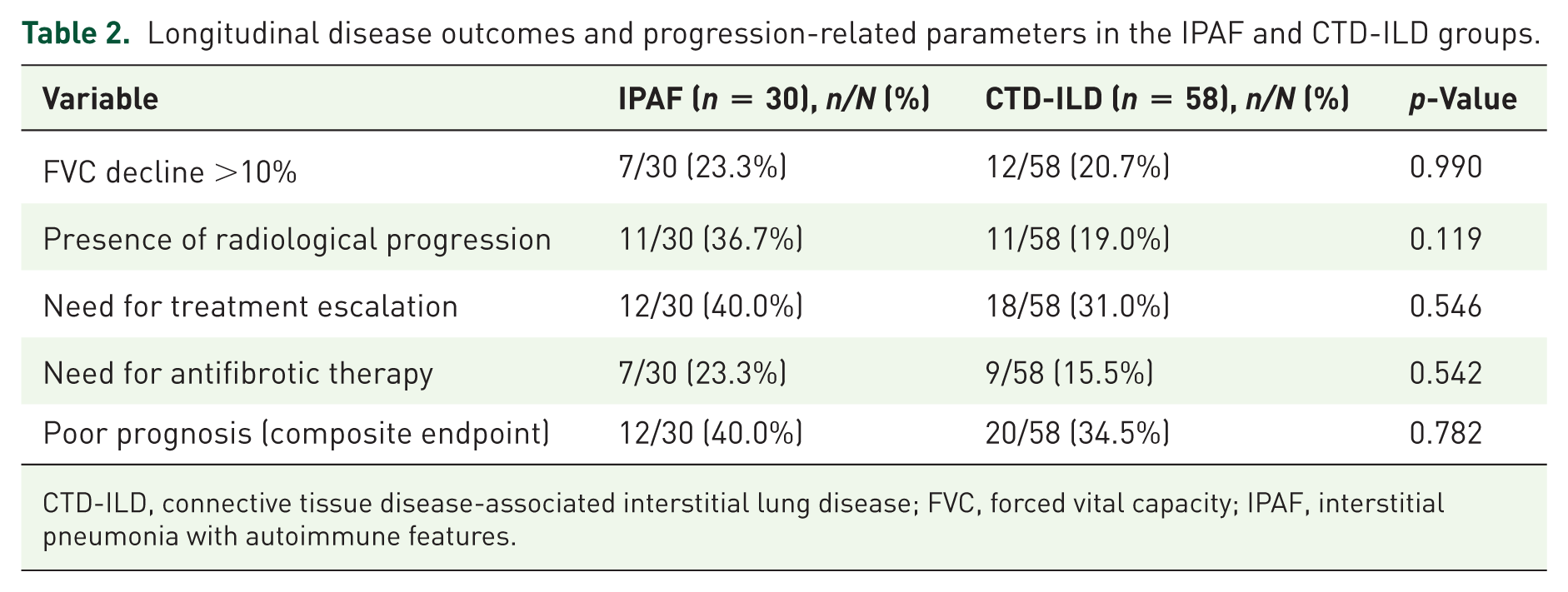
CTD-ILD, connective tissue disease-associated interstitial lung disease; FVC, forced vital capacity; IPAF, interstitial pneumonia with autoimmune features.
As demonstrated in Table 3, persistent ESR elevation and persistent CRP elevation were significantly associated with an increased risk of poor prognosis in univariable analysis.
Baseline predictors associated with poor prognosis: univariable analysis.
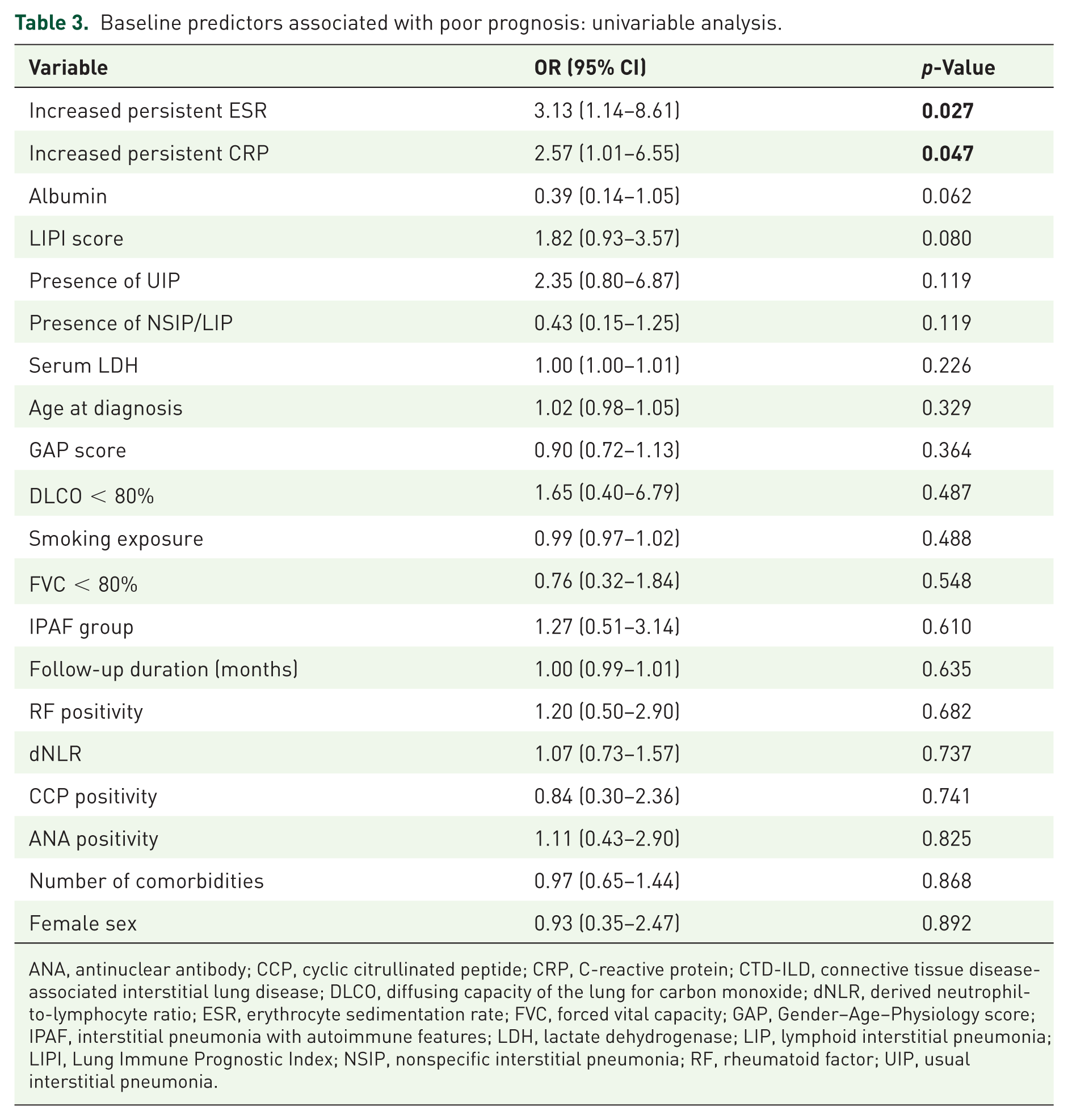
ANA, antinuclear antibody; CCP, cyclic citrullinated peptide; CRP, C-reactive protein; CTD-ILD, connective tissue disease-associated interstitial lung disease; DLCO, diffusing capacity of the lung for carbon monoxide; dNLR, derived neutrophil-to-lymphocyte ratio; ESR, erythrocyte sedimentation rate; FVC, forced vital capacity; GAP, Gender–Age–Physiology score; IPAF, interstitial pneumonia with autoimmune features; LDH, lactate dehydrogenase; LIP, lymphoid interstitial pneumonia; LIPI, Lung Immune Prognostic Index; NSIP, nonspecific interstitial pneumonia; RF, rheumatoid factor; UIP, usual interstitial pneumonia.
As shown in Table 4, the exclusion of patients with UIP-pattern disease did not substantially alter the overall outcome comparisons between the IPAF and CTD-ILD groups.
Sensitivity analysis of longitudinal outcomes in patients without a UIP pattern: comparison between non-UIP IPAF and non-UIP CTD-ILD groups.
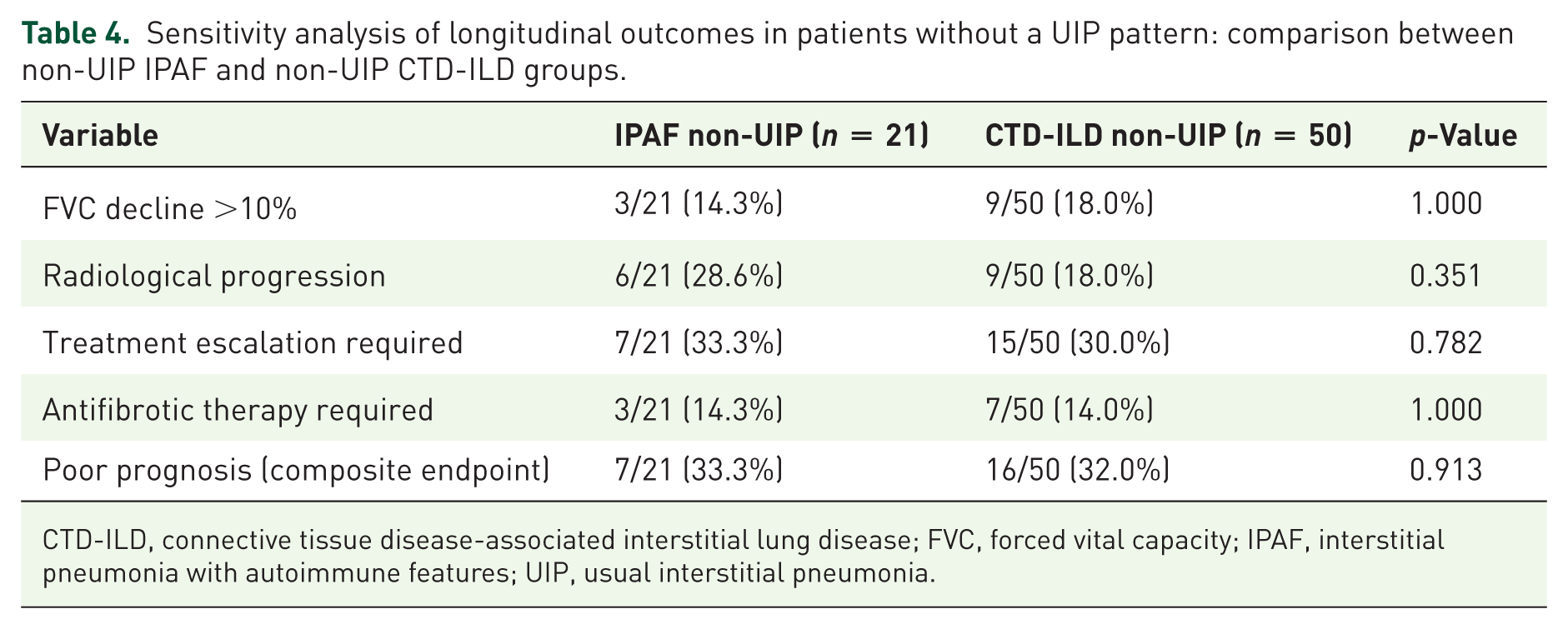
CTD-ILD, connective tissue disease-associated interstitial lung disease; FVC, forced vital capacity; IPAF, interstitial pneumonia with autoimmune features; UIP, usual interstitial pneumonia.
Outcome analyses stratified according to persistent ESR elevation and UIP-pattern status are summarized in Table 5. Persistent ESR elevation identified a clearly higher-risk subgroup, with significantly increased rates of poor prognosis (57.1% vs 29.9%, p = 0.023), treatment escalation (52.4% vs 28.4%, p = 0.043), and antifibrotic therapy requirement (33.3% vs 13.4%, p = 0.039) compared to patients without ESR elevation. In contrast, although a UIP pattern was associated with greater functional decline (41.2% vs 16.9%, p = 0.029) and higher antifibrotic use (35.3% vs 14.1%, p = 0.042), it did not translate into a statistically significant difference in the composite poor prognosis outcome (52.9% vs 32.4%, p = 0.114).
Comparative outcome analysis according to persistent ESR elevation and radiological UIP pattern status.
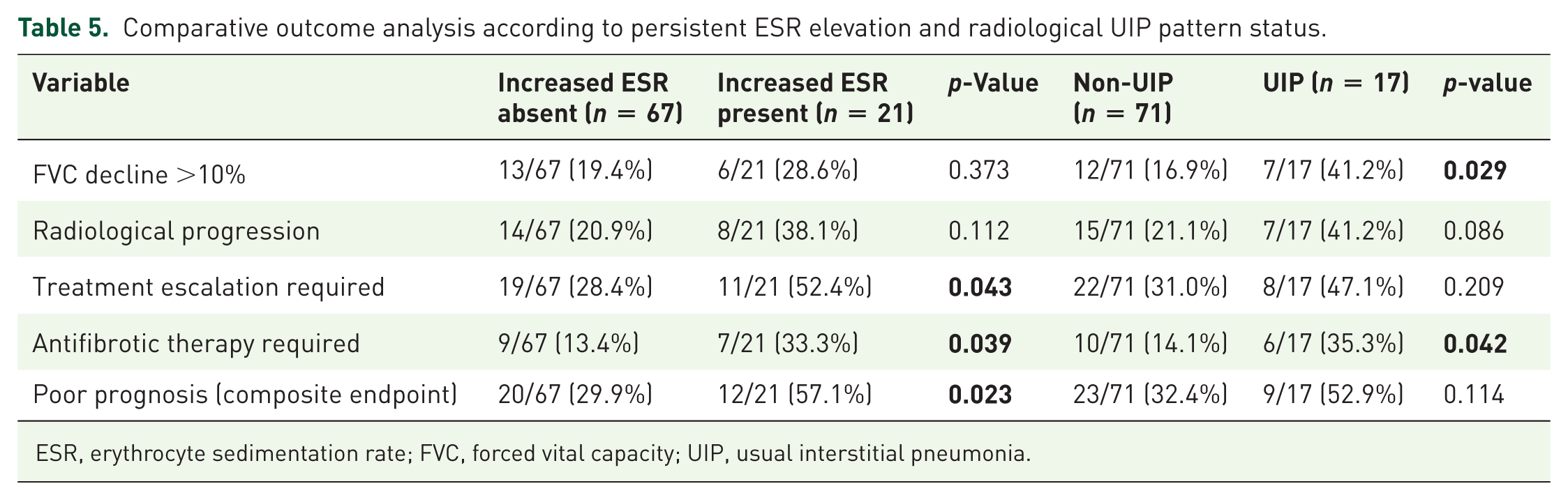
ESR, erythrocyte sedimentation rate; FVC, forced vital capacity; UIP, usual interstitial pneumonia.
As shown in Table 6, persistent ESR elevation remained an independent predictor of poor prognosis after multivariable adjustment. In the multivariable model, the presence of persistent ESR elevation persisted as an independent predictor of poor prognosis. Patients with persistent ESR positivity had approximately a 3.25-fold higher likelihood of experiencing an adverse outcome. Following exclusion of patients with a UIP pattern, no statistically significant differences were detected between the IPAF and CTD-ILD groups with respect to the composite poor prognosis endpoint or its individual components. Within the non-UIP cohort, persistent elevation of ESR remained an independent predictor of adverse outcome (odds ratio 3.79, 95% CI 1.03–13.88; p = 0.045).
Parsimonious multivariable model adjusted for key covariates.
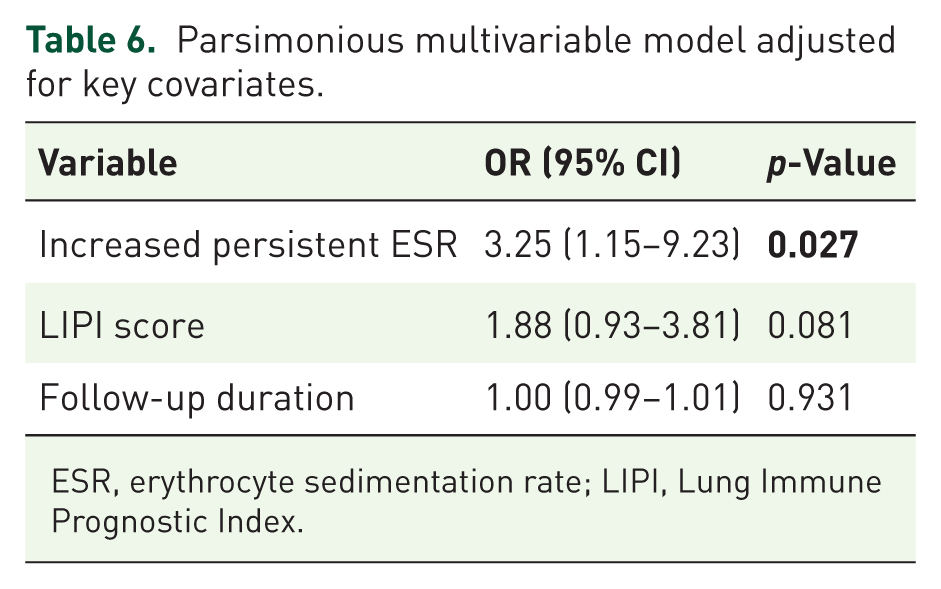
ESR, erythrocyte sedimentation rate; LIPI, Lung Immune Prognostic Index.
The discriminatory performance of GAP score, LIPI score, and dNLR for predicting poor prognosis is illustrated in Figure 2. ROC analysis demonstrated poor overall discriminative performance for all evaluated scores (AUC values < 0.60), indicating limited ability to reliably distinguish patients at higher risk of adverse outcomes. LIPI demonstrated high specificity but low sensitivity, suggesting it may identify a small high-risk subgroup but is not suitable as a general screening tool. For dNLR, a cut-off around 2.1 yielded moderate sensitivity and specificity.

Receiver operating characteristic (ROC) analysis of prognostic scores for predicting poor prognosis in patients with ILD.
Overall, the findings consistently indicate that there is no meaningful difference in poor prognosis between IPAF and CTD-ILD, even after accounting for potential confounders such as radiologic pattern. While the presence of a UIP pattern was linked to greater functional decline and increased antifibrotic therapy requirements, it did not independently translate into worse composite outcomes. Notably, persistent elevation of ESR emerged as the most robust and consistent predictor of adverse prognosis.
Discussion
In our cohort, no significant difference in adverse prognosis was observed between IPAF and CTD-ILD, and this finding remained consistent even after excluding patients with UIP. Collectively, these results suggest that disease trajectory and progression dynamics may play a more decisive role than diagnostic classification alone. In contrast to our findings, prior reports have indicated that certain clinical factors may influence prognosis in IPAF. For instance, older age has been identified as an adverse prognostic indicator in this population. 14 In contrast, although advanced age and higher GAP scores were more frequently observed in patients with IPAF, the composite outcome reflecting overall prognosis remained comparable between the two groups. The comparable outcome rates between groups should be interpreted cautiously, as the shorter follow-up duration in the IPAF cohort may have limited the opportunity to capture delayed progression events, in the context of unequal follow-up duration.
Persistent inflammation emerged as the central signal in our analysis, with sustained elevation of the ESR representing the most robust independent predictor of adverse outcomes, while the effect of CRP attenuated after multivariable adjustment. This pattern points to a closer link between chronic inflammatory burden and fibrotic progression rather than transient inflammatory activity. Consistent with our results, prior studies have shown that neutrophil-to-lymphocyte ratio and systemic immune-inflammation index are elevated in both CTD-ILD and IPF compared to controls, reflecting an increased systemic inflammatory burden. 15 Besides, elevated IL-6 levels in patients with RA-ILD, compared to those without ILD, and their positive correlation with disease activity suggest a potential role of IL-6-mediated inflammation in the pulmonary involvement. 16 These findings can be interpreted in light of shared pathogenic mechanisms linking inflammation and fibrosis across ILD subtypes. Our results, particularly the association with persistent inflammation, support the concept of an inflammation–immunity–fibrosis continuum in CTD-ILD. Ongoing immune activation—driven by sustained T-cell responses and B-cell dysregulation—maintains chronic inflammatory signaling, which in turn promotes progressive fibrotic remodeling through interconnected cytokine pathways. 17 Another point in which our findings align with the existing literature is the predominance of female patients in the CTD-ILD group. Previous comparative studies have consistently associated CTD-ILD with female sex and a higher prevalence of autoantibody positivity—particularly anti-CCP antibodies such as in our cohort. 18
Findings that diverged from the existing literature were also observed. The radiological evidence of fibrosis on HRCT—particularly the presence of a UIP pattern—has been linked to an increased likelihood of early disease progression in IPAF. 19 In addition, older age and the presence of the UIP pattern were related to poorer prognosis in IPAF. 20 Our results pointed in a different direction. In our cohort, UIP was primarily linked to a steeper decline in FVC and a greater need for antifibrotic therapy, underscoring its association with a more aggressive fibrotic phenotype, rather than consistently translating into a clearly worse overall prognosis. This apparent discrepancy likely reflects the heterogeneous nature of the composite endpoint, in which individual components are not uniformly driven by the same underlying mechanisms. In contrast, persistent systemic inflammation, as indicated by elevated ESR, appears to exert a broader and more integrated effect across multiple domains of disease activity. These findings suggest that fibrotic architecture and inflammatory burden represent related but distinct processes, with ongoing inflammation potentially serving as a more comprehensive determinant of overall disease trajectory.
Treatment approaches varied across groups, with MMF used more frequently in IPAF, while cyclophosphamide and rituximab were preferentially employed in CTD-ILD. Such heterogeneity in therapeutic strategies may have tempered observable differences in outcomes, potentially masking intrinsic biological disparities. Importantly, treatment allocation was not randomized and likely mirrored baseline disease phenotype and severity, which should be carefully considered when interpreting comparative results. In this context, the treatment patterns observed in our cohort are broadly aligned with existing literature and reflect contemporary clinical practice across ILD subtypes. Immunomodulatory therapy remains a cornerstone in CTD-ILD management, in contrast to IPF, where its role is limited. Current paradigms increasingly support a combined therapeutic approach that targets both inflammatory and fibrotic pathways, integrating immunosuppressive agents with antifibrotic therapies. 21 In routine clinical care, most patients receive corticosteroids alongside steroid-sparing agents such as MMF, whereas biologic therapies—particularly rituximab—are reserved for selected cases with more severe or refractory disease. Rituximab, an anti-CD20 monoclonal antibody, exerts its effect through B-cell depletion, targeting key pathogenic mechanisms in CTD-ILD including autoantibody production, antigen presentation, cytokine release, and ectopic lymphoid structure formation. Through these pathways, it modulates both immune activation and downstream fibrotic processes. Accumulating evidence suggests that rituximab may stabilize or even improve pulmonary function, supporting its role as a valuable therapeutic option in CTD-ILD. 22 More recently, antifibrotic therapies have gained increasing attention, particularly in patients exhibiting progressive fibrotic phenotypes. 23 Prior studies indicate that antifibrotic agents can be safely administered in IPAF and may contribute to slowing FVC decline. 24 Within our cohort, MMF and rituximab emerged as the most commonly utilized agents in IPAF and CTD-ILD, respectively. Additionally, antifibrotic therapy with nintedanib was initiated in 7 of 30 patients and was generally well tolerated among those treated. Collectively, these findings highlight not only the evolving complexity of therapeutic strategies in ILD but also the challenge of disentangling treatment effects from underlying disease biology in observational cohorts.
These findings suggest that GAP, LIPI, and dNLR should not be considered standalone prognostic tools in autoimmune-related ILD, although they may still provide complementary information when interpreted alongside clinical, functional, and radiological assessment. The modest performance of these indices may reflect the biological heterogeneity of IPAF and CTD-ILD, where single composite scores are unlikely to capture the complex interplay between inflammation and fibrosis. From a practical standpoint, IPAF patients should be monitored with a level of vigilance comparable to CTD-ILD, with particular attention to persistently elevated ESR as a dynamic marker to be integrated into follow-up and therapeutic decision-making. Finally, the presence of a UIP pattern seems to inform treatment intensity rather than independently define overall prognosis.
Limitations
Several limitations should be considered when interpreting these findings. First, the retrospective design precludes causal inference and is inherently vulnerable to selection and information bias. The single-center setting further limits external validity, as the cohort—drawn from a tertiary referral center—was likely enriched for more complex or treatment-refractory cases, thereby constraining generalizability to broader, community-based ILD populations and different healthcare contexts.
Second, the overall sample size was modest, particularly within the IPAF subgroup, which may have reduced statistical power and increased the risk of type II error. This limitation may have hindered the detection of subtle but clinically relevant differences between groups.
Third, there was a notable imbalance in follow-up duration (24 vs 48 months), which may have introduced ascertainment bias and influenced the evaluation of progression-related outcomes. The shorter follow-up in IPAF patients, in particular, may have limited the assessment of long-term disease trajectories and potentially obscured between-group differences.
Fourth, treatment strategies were heterogeneous and not protocol-driven but rather based on clinician judgment in a real-world setting. This introduces the possibility of confounding by indication, as treatment decisions were likely influenced by baseline disease severity and clinical phenotype. Moreover, because treatment escalation was incorporated into the composite outcome, adjustment for treatment exposure within prognostic models was inherently constrained, complicating causal interpretation.
Fifth, although the use of a composite endpoint allowed a broader capture of disease progression, variability in its components and the absence of external validation necessitate cautious interpretation of the results.
Finally, histopathological confirmation was limited, restricting a more granular understanding of underlying disease mechanisms and reducing the ability to correlate clinical outcomes with specific pathological patterns. Collectively, these limitations underscore the need for prospective, multicenter studies with standardized treatment approaches and validated outcome measures to better delineate prognostic determinants in ILD.
Key strengths of this study include the use of real-world data reflecting routine clinical practice, enhancing its practical relevance, and a direct comparative design between IPAF and CTD-ILD, which allows for clinically meaningful contextualization of outcomes. The inclusion of detailed UIP-based analyses, along with subgroup evaluations, provides additional granularity in understanding disease behavior across fibrotic phenotypes. Furthermore, the application of multivariable modeling strengthens the analytical framework by enabling adjustment for potential confounders and supporting more robust identification of independent prognostic factors.
Conclusion
Rather than diagnostic categorization alone, the persistence of inflammatory activity appears to be a more decisive driver of prognosis. In our cohort, sustained elevation of the ESR was the only variable independently associated with adverse outcomes, underscoring the potential role of chronic systemic inflammation in shaping disease trajectory in both IPAF and CTD-ILD. While these observations highlight ESR as a clinically informative signal, confirmation in larger prospective studies is required before it can be adopted as a routine prognostic tool.
Supplemental Material
sj-docx-1-tar-10.1177_17534666261465583 – Supplemental material for Inflammatory activity as a predictor of clinical deterioration in interstitial pneumonia with autoimmune features: a comparative cohort study with connective tissue disease-associated interstitial lung disease
Supplemental material, sj-docx-1-tar-10.1177_17534666261465583 for Inflammatory activity as a predictor of clinical deterioration in interstitial pneumonia with autoimmune features: a comparative cohort study with connective tissue disease-associated interstitial lung disease by Firdevs Ulutaş, Nilüfer Yiğit, Gülay Güngör and Veli Çobankara in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The authors would like to acknowledge the contributions of all clinicians and healthcare professionals involved in the multidisciplinary evaluation and management of the patients included in this study. Their clinical expertise and collaborative efforts were essential to the integrity and completeness of the dataset.
Declarations
Artificial intelligence declaration
Generative artificial intelligence (AI) tools, including ChatGPT (OpenAI), were used solely to support language refinement, grammatical correction, structural organization, and enhancement of overall readability during manuscript preparation. No AI tools were used for data generation, statistical analysis, data interpretation, or independent scientific reasoning. All scientific content, methodological decisions, analyses, and conclusions were developed exclusively by the authors. The authors assume full responsibility for the accuracy, originality, and integrity of the manuscript and confirm that all outputs generated with AI assistance were carefully reviewed, critically evaluated, and appropriately revised to ensure compliance with academic and ethical standards. During the preparation of this manuscript, ChatGPT (OpenAI) was used solely for language editing, grammatical refinement, literature review. The authors reviewed and edited all content critically and take full responsibility for the accuracy and integrity of the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.