
Abstract
Parkinson’s disease (PD) remains the second most common neurodegenerative disorder globally, yet disease-modifying therapies remain elusive. The past decade of therapeutic development has been dominated by the hypothesis that alpha-synuclein aggregates spread in a prion-like manner. This catalysed the development of immunotherapy approaches targeting extracellular alpha-synuclein. However, high-profile Phase II and III clinical trials, such as those for cinpanemab and prasinezumab, failed to demonstrate clinical efficacy. This narrative review evaluates alpha-synuclein-targeted immunotherapies, their mechanistic rationale, the underlying reasons for recent clinical trial failures and to propose alternative metabolic frameworks. A literature search of peer-reviewed articles published up to 10 December 2025 was performed using the PubMed database to evaluate immunotherapies, mechanistic models and trial failures in PD. The prevailing ‘prion-like’ spread hypothesis may be overemphasised in the context of human disease. We propose the ‘Single-Neuron Degeneration Hypothesis’, suggesting that PD pathology is primarily driven by intracellular autotoxicity – specifically involving the dopamine metabolite aminochrome, mitochondrial dysfunction and oxidative stress – occurring independently within each dopaminergic neuron. Multifactorial contributors to trial failure include the inadequacy of preclinical models such as MPTP, the insensitivity of the MDS-UPDRS, diagnostic heterogeneity and the statistical reality of neuronal loss rates. Future therapeutic strategies must integrate intracellular neuroprotection alongside extracellular clearance approaches, with significantly earlier intervention.
Plain language summary
Parkinson’s disease is a common brain disorder, yet we still lack treatments that can slow its progression. For years, scientists believed the disease spread like an infection, with a toxic protein moving from one brain cell to another. This theory led to massive clinical trials testing "antibodies" designed to catch this protein in the space between cells. Unfortunately, these major trials have failed to show clinical benefits. This review explains why those trials likely missed the mark. First, the damage mostly happens inside the brain cells, where these large antibody treatments cannot reach. Second, the "spread" theory may be overemphasized. We propose a "Single-Neuron" theory: brain cells die individually because they can no longer neutralize their own internal toxic byproducts, such as "aminochrome". Furthermore, humans lose brain cells very slowly in Parkinson’s—only about 60 per day. Current medical tests are simply not sensitive enough to measure such a tiny change over a standard one-year study. To fix this, future research must shift focus. We should develop drugs that "armor" the cell from the inside by boosting its natural protective systems (specifically a pathway called NRF2). We also need more precise brain scans and better patient selection to ensure we are testing the right treatments at the right time. By protecting cells from their own internal metabolic "fire," we may finally find a way to slow down this disease.
Keywords
Introduction
Parkinson’s disease (PD) is characterised clinically by the tetrad of bradykinesia, resting tremor, rigidity and postural instability, 1 and pathologically by the progressive loss of dopaminergic neurons in the substantia nigra pars compacta coupled with the presence of Lewy bodies composed primarily of aggregated alpha-synuclein – first described in detail by Spillantini and colleagues. 2 While the exact primary neurotoxin responsible for this cell loss remains debated, the scientific community broadly agrees that factors such as the generation of neurotoxic alpha-synuclein oligomers, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, defects in lysosomal and proteasomal protein breakdown systems and neuroinflammation are contributing elements to neuronal death.3–11
For years, the gold standard of therapeutic research has focused on the accumulation of alpha-synuclein. The discovery that alpha-synuclein fibrils could induce aggregation in neighbouring cells in vitro and in animal models gave rise to the ‘prion-like spread’ hypothesis.12–20 This theory posits that pathological alpha-synuclein acts as a seed, escaping a dying neuron to infect healthy neighbours, thereby driving the progression of the disease through the brain’s connectome. The rationale for targeting alpha-synuclein is grounded in pivotal studies demonstrating its pathogenicity. Transgenic mouse models overexpressing human alpha-synuclein develop hallmark motor deficits and neuronal inclusions. 21 It is also important to note that numerous interventions targeting alpha-synuclein aggregates and the aggregation process itself have shown great promise in cellular and animal models, further supporting the role of toxic aggregates in PD aetiology. Furthermore, in vivo studies have shown that inoculating seeding-competent alpha-synuclein fibrils into the brain accelerates the spread of pathology and neuronal death.22,23 These findings provided the translational basis for the hypothesis that neutralising extracellular alpha-synuclein could arrest disease progression.
Based on this premise, the pharmaceutical industry invested heavily in immunotherapy. The logic appeared sound: if the disease progresses via the extracellular transit of toxic proteins, then sequestering these proteins with antibodies should halt disease progression. However, the recent failures of major antibody trials have cast serious doubt on this approach as a standalone strategy. Importantly, ongoing advances in receptor-mediated transcytosis engineering and targeted protein degradation continue to refine immunotherapy, and the field should not abandon this avenue entirely. This narrative review explores the multi-dimensional causes of these failures, ranging from molecular misconceptions regarding the primary driver of toxicity to flaws in clinical trial design and execution, and proposes complementary intracellular therapeutic strategies.
Search strategy
For this narrative review, we performed a non-systematic literature search of peer-reviewed articles published in English up to 10 December 2025, primarily using the PubMed database. Search terms were designed to capture studies relevant to alpha-synuclein–targeted immunotherapies in PD, their mechanistic rationale, and reasons for clinical trial failure. These included combinations of: (‘Parkinson*’ OR ‘idiopathic Parkinson’s disease’) AND (‘alpha-synuclein’ OR ‘α-synuclein’), together with (‘monoclonal antibody’ OR ‘immunotherapy’ OR ‘passive immunization’ OR ‘clinical trial’ OR ‘Phase 2’ OR ‘Phase 3’). Additional terms were used to identify mechanistic and translational literature, including (‘prion-like spread’ OR ‘protein propagation’), (‘oligomer*’ OR ‘aggregation’), (‘neuroprotection’), (‘mitochondrial dysfunction’ OR ‘oxidative stress’), (‘aminochrome’ OR ‘dopamine oxidation’), (‘MPTP’ OR ‘preclinical model’) and (‘UPDRS’ OR ‘clinical outcome measures’).
Given the focus on understanding therapeutic failure, both preclinical and clinical studies were included, as well as publications addressing trial design, outcome measures, disease heterogeneity and alternative pathogenic hypotheses. Additional relevant articles were identified through manual screening of reference lists from key clinical trials, reviews and mechanistic studies. This is a narrative review designed to synthesise the conceptual gap between immunotherapy failures and metabolic degeneration. A systematic review was not conducted as the aim was to integrate disparate mechanistic hypotheses rather than quantify trial outcomes.
Clinical studies on neuroprotection
The most significant advance in the pharmacological management of PD was the introduction of L-dopa in 1967. L-dopa is highly effective, enabling patients to regain many lost motor functions once symptoms have manifested. Motor symptoms appear when approximately 60–70% of substantia nigra cells and 70%–80% of dopamine levels in striatal nuclei have been lost.24–26 Importantly, L-dopa is a palliative, symptomatic treatment; it does not halt or reverse PD progression. The primary complication with this treatment is the development of severe secondary symptoms, notably dyskinesia, as well as debilitating motor and non-motor fluctuations, all of which occur after 4–6 years of therapy and severely diminish patients’ quality of life.
Based on the hypothesis that oxidative stress plays a key role in the degeneration of dopaminergic neurons, several clinical trials have been conducted using antioxidants. Evidence of oxidative stress in PD patients has included elevated levels of oxidised glutathione, reduced glutathione peroxidase activity and high levels of malondialdehyde in blood samples. 19 Furthermore, reduced expression levels of antioxidant enzymes, such as catalase, superoxide dismutase and glutathione peroxidase, have been observed in PD patients. However, as discussed in the following section, the failure of these antioxidant trials shaped the field’s subsequent transition toward alpha-synuclein-targeted antibody therapies, which focused on aggregation and propagation. In retrospect, the failure of these antioxidants may have occurred in part because they did not specifically target the dopaminergic neuron’s intracellular vulnerability to aminochrome, rather than providing generalised antioxidant support.
Coenzyme Q (CoQ) was employed in clinical studies based on the premise that mitochondrial dysfunction is a fundamental factor in the degenerative process of the nigrostriatal system in PD. 27 Studies observed decreased activity of complexes I and II in the mitochondria of PD patients, which correlated with a reduction in CoQ levels.27–32 Preclinical findings showed neuroprotective effects of coenzyme Q10 on tyrosine hydroxylase activity and bioenergetic status, and it was observed to slow the progression of motor deterioration in PD patients at high concentrations in dose-determination studies.30–33 Coenzyme Q10 also protected against mitochondrial depolarisation, apoptosis and cell death induced by rotenone. 34
Mitoquinone, a powerful antioxidant analogue of coenzyme Q, also demonstrated a clear neuroprotective effect in cell culture models treated with the neurotoxin MPP+. In MPTP-treated mice, mitoquinone reversed the loss of motor activity and dopaminergic neurons and prevented the inactivation of aconitase. 35 Urate, a potent antioxidant and the final product of purine metabolism, was also utilised in clinical trials. 36 Studies with rats showed that urate decreased motor deficits, reduced the loss of dopaminergic neurons and increased levels of reduced glutathione.37,38 Urate levels in cerebrospinal fluid and plasma have been associated with a lower risk of developing PD. 39
The discovery of iron deposits in the postmortem brains of PD patients led some researchers to suggest that these deposits might be a source of oxidative stress, as ferrous iron catalyses the Fenton reaction to generate hydroxyl radicals. Iron chelators, such as deferiprone, were shown in preclinical studies to protect cells against lipoperoxidation and promote the emergence of new tyrosine hydroxylase-positive processes in lesioned dopaminergic neurons.40,41 Regrettably, clinical trials evaluating CoQ, mitoquinone, urate and deferiprone all failed to show that these antioxidants could modify the disease course or provide a benefit in the treatment of PD.9,42–46 The reasons for these failures were multifactorial, including insufficient target specificity, unintended effects on cellular homeostasis and signalling, and limitations of outcome measures – issues that, as elaborated below, also plagued the subsequent alpha-synuclein antibody trials.
Alpha-synuclein biology and the multiple-species problem
Alpha-synuclein is a 140-amino acid protein that typically exists in a monomeric form. 47 Although its exact physiological function is still being elucidated, it is believed to participate in presynaptic vesicle trafficking and membrane regulation. However, under specific conditions, alpha-synuclein can adopt multiple misfolded conformations, generating a spectrum of species – monomers, oligomers, protofibrils and mature fibrils – each with distinct biochemical properties and degrees of neurotoxicity. This conformational diversity is directly relevant to the limited efficacy of antibody strategies: different antibodies may preferentially bind one species (e.g., aggregated fibrils) while poorly targeting the most neurotoxic conformers (e.g., small, soluble oligomers), potentially explaining why antibodies that achieve target engagement in terms of fibril clearance nonetheless fail to confer clinical benefit.
The link between alpha-synuclein and PD was solidified when mutations like A30P and A53T were linked to hereditary forms of the disease. When alpha-synuclein forms fibrils, it constitutes the deposits known as Lewy bodies. Critically, wild-type alpha-synuclein also forms oligomers; the familial mutations merely accelerate this process to a somewhat greater extent and may generate oligomers with slightly different structural properties. The progression of PD through various brain regions, as described in the Braak staging model, has been proposed to be driven by the spread of Lewy pathology, leading some researchers to suggest prion-like behaviour for alpha-synuclein.48,49 Braak staging of post-mortem brain tissue – carried out routinely by brain banks as standard practice – demonstrates that the severity of PD symptoms correlates with the extent of Lewy body pathology, progressing from the brainstem through limbic and neocortical regions. This staging is based not merely on in vitro or animal data, but on extensive post-mortem human tissue analysis, and represents strong evidence for pathology spreading through interconnected neural circuits. The proposed single-neuron model does not fully account for this stereotyped, network-based propagation and any alternative framework must grapple with this evidence.
Nevertheless, the function of Lewy bodies in the spread and progression of idiopathic PD (iPD) remains contentious: they are found in surviving neurons in post-mortem studies; certain familial PD cases, such as those associated with LRRK2 gene mutations, can develop without Lewy bodies (though not all LRRK2-PD cases are Lewy body–negative50,51); and some researchers propose a potential neuroprotective role for Lewy bodies, suggesting they may sequester toxic oligomers. Conversely, alpha-synuclein oligomers exert neurotoxic effects through several distinct mechanisms: they cause mitochondrial damage by directly impairing Complex I and increasing reactive oxygen species (ROS)52–55; they lead to proteasomal dysfunction by blocking substrate entry; they trigger ER stress55,56; they cause synaptic impairment by interfering with SNARE complex formation; they generate oxidative stress by promoting ROS and lipid peroxidation; and they trigger neuroinflammation by activating glial cells.57–64 Furthermore, prefibrillar oligomers accumulate in the ER, triggering the Unfolded Protein Response (UPR), which ultimately activates apoptotic pathways.65–67 Crucially, these intracellular cascades – mitochondrial collapse and ER stress – likely occur upstream of the extracellular spread targeted by antibodies.
In familial PD, specific gene mutations – such as point mutations or multiplications of the SNCA gene – directly drive the misfolding and formation of neurotoxic oligomers. In sporadic idiopathic PD (iPD), however, these causative mutations are absent. While age-related decline in proteostasis and environmental factors is broadly implicated, they lack the specificity needed to explain the precise vulnerability of dopaminergic neurons.68–72 As explored later in this review, we argue that the endogenous accumulation of aminochrome – a specific, toxic byproduct of dopamine oxidation – serves as this missing biochemical trigger, initiating alpha-synuclein oligomerisation in iPD even in the absence of genetic abnormalities.
The reality check: Alpha-synuclein antibody failures
The clinical landscape has recently been marked by disappointing results from large-scale trials. For a detailed account of the trial design issues that contributed to these failures, see the ‘Flaws in Clinical Trials and Preclinical Modelling’ section below. Both Roche’s prasinezumab (PASADENA study) and Biogen’s cinpanemab (SPARK study) failed to meet their primary endpoints in slowing the progression of motor symptoms as measured by the MDS-UPDRS.73,74 These antibodies were designed to bind aggregated alpha-synuclein in the extracellular space, facilitating its clearance by microglia and preventing uptake by adjacent neurons.
These antibodies should theoretically also mitigate neuroinflammation by preventing the activation of microglia by extracellular aggregates. However, full-sized monoclonal antibodies have poor intracellular penetration. Consequently, they cannot neutralise the intraneuronal oligomers driving mitochondrial and ER toxicity – cascades that occur upstream of the extracellular spread these agents were designed to intercept.
The failure of these agents confronts the field with two possibilities: either the antibodies did not achieve sufficient target engagement in the brain, or the target itself – extracellular alpha-synuclein spreading – is not the primary driver of disease progression during the symptomatic phase of PD. Biomarker data from both the PASADENA and SPARK trials suggest that target engagement was achieved to a meaningful extent, as evidenced by reductions in cerebrospinal fluid alpha-synuclein species following treatment.73,74 These findings oblige us to seriously consider that the therapeutic hypothesis itself – while scientifically valid – may be insufficient as a standalone strategy. Importantly, this does not invalidate the prion-like spread model entirely; rather, it suggests that extracellular clearance alone may not be enough when intracellular autotoxic processes are already well underway.
It is important to acknowledge that the field continues to evolve. Two major ongoing developments may overcome the limitations of first-generation antibody therapies: (1) engineering antibodies for enhanced blood–brain barrier (BBB) crossing, exploiting receptor-mediated transcytosis either by directly engineering the mAbs or by encapsulating them in liposomes targeting the transferrin receptor; and (2) the use of targeted protein degradation approaches to more effectively eliminate extracellular alpha-synuclein aggregates. These advances underscore the importance of pursuing – and understanding – all available avenues.
The single-neuron degeneration hypothesis: An alternative paradigm
The ‘Single-Neuron Degeneration Hypothesis’ revisits the classical concept of cell-autonomous death, 75 offering a necessary counter-narrative to the currently dominant prion-like spread theory. It is important to clarify that this is not a novel discovery: the concept of cell-autonomous neurodegeneration driven by intracellular dopamine metabolites – including aminochrome – has been developed principally by Huenchuguala and Segura-Aguilar, 75 upon whose foundational work this synthesis draws. The contribution of the present review is to argue for the re-prioritisation of this established intracellular model in the context of clinical trial design and to integrate it with the specific failure modes of recent antibody trials. We propose that dopaminergic neurons die individually due to an inability to manage endogenous toxic byproducts – rather than through neighbour-to-neighbour infection – and that this model better explains the extremely slow progression of iPD.76–80
We acknowledge, however, that this model in its current form does not provide a complete account of PD propagation. The Braak model – supported by extensive post-mortem human tissue data – provides compelling evidence for spatially ordered spread through interconnected brain circuits, most likely via extracellular aggregates. The single-neuron hypothesis posits isolated cell deaths without an explicit mechanism for this network-based progression. Future work must reconcile these two frameworks; they may be complementary rather than mutually exclusive, with intracellular metabolic erosion as the initiating event and secondary extracellular spreading amplifying the pathology.
While the toxic roles of intracellular mechanisms like aminochrome81–83 and oxidative stress are well-documented, recent therapeutic strategies have largely ignored them in favour of extracellular targets. We propose refocusing on these established intracellular pathways. This hypothesis is consistent with the observation that an external, expansive neurotoxin would likely cause a synchronous loss of all affected and adjacent neurons, leading to rapid degeneration rather than the slow, decades-long decline observed in iPD.
In this model, DOPAL (3,4-dihydroxyphenylacetaldehyde) – the aldehyde metabolite of dopamine generated by monoamine oxidase – is also considered as a candidate neurotoxin. DOPAL is a highly reactive catecholaldehyde that covalently modifies proteins, promotes alpha-synuclein oligomerisation and has been detected in post-mortem PD brains. However, DOPAL is excluded as the primary neurotoxin in the single-neuron model because evidence suggests it can be transmitted to adjacent cells and can oligomerise alpha-synuclein in human glial cells, 84 which would endow it with expansive properties inconsistent with strictly cell-autonomous degeneration.
The chemistry of autotoxicity: Aminochrome
The neurotransmitter dopamine is a double-edged sword. Dopaminergic neurons in the nigrostriatal system normally synthesise neuromelanin, a process involving the oxidation of dopamine into transient ortho-quinones.85,86 Among known endogenous neurotoxins, aminochrome is the most stable and neurotoxic ortho-quinone generated during this synthesis.81,82
In a healthy neuron, this synthesis is rendered harmless by robust defence mechanisms. Enzymes such as DT-diaphorase (NQO1) and glutathione transferase M2-2 (GSTM2) neutralise aminochrome. 85 Astrocytes contribute to this protection by releasing exosomes containing GSTM2, which are absorbed by dopaminergic neurons. However, in the degenerating neuron characteristic of iPD, these mechanisms fail. Persistent blockade of DT-diaphorase leads to the formation of neurotoxic alpha-synuclein oligomers, suggesting that aminochrome is the endogenous neurotoxin that initiates these harmful oligomers in iPD.86–89
Aminochrome – formed during neuromelanin synthesis – induces oxidative stress, alpha-synuclein oligomer formation, mitochondrial dysfunction by impairing Complex I, and neuroinflammation.81–83 Crucially, aminochrome is rapidly reduced or forms protein adducts inside the neuron immediately after formation, which prevents its excretion and spread to neighbouring cells. 80 This characteristic makes aminochrome a strong candidate for the single-neuron degeneration model in iPD. Figure 1 illustrates the contrast between a healthy dopaminergic neuron and one undergoing the proposed single-neuron degeneration process.
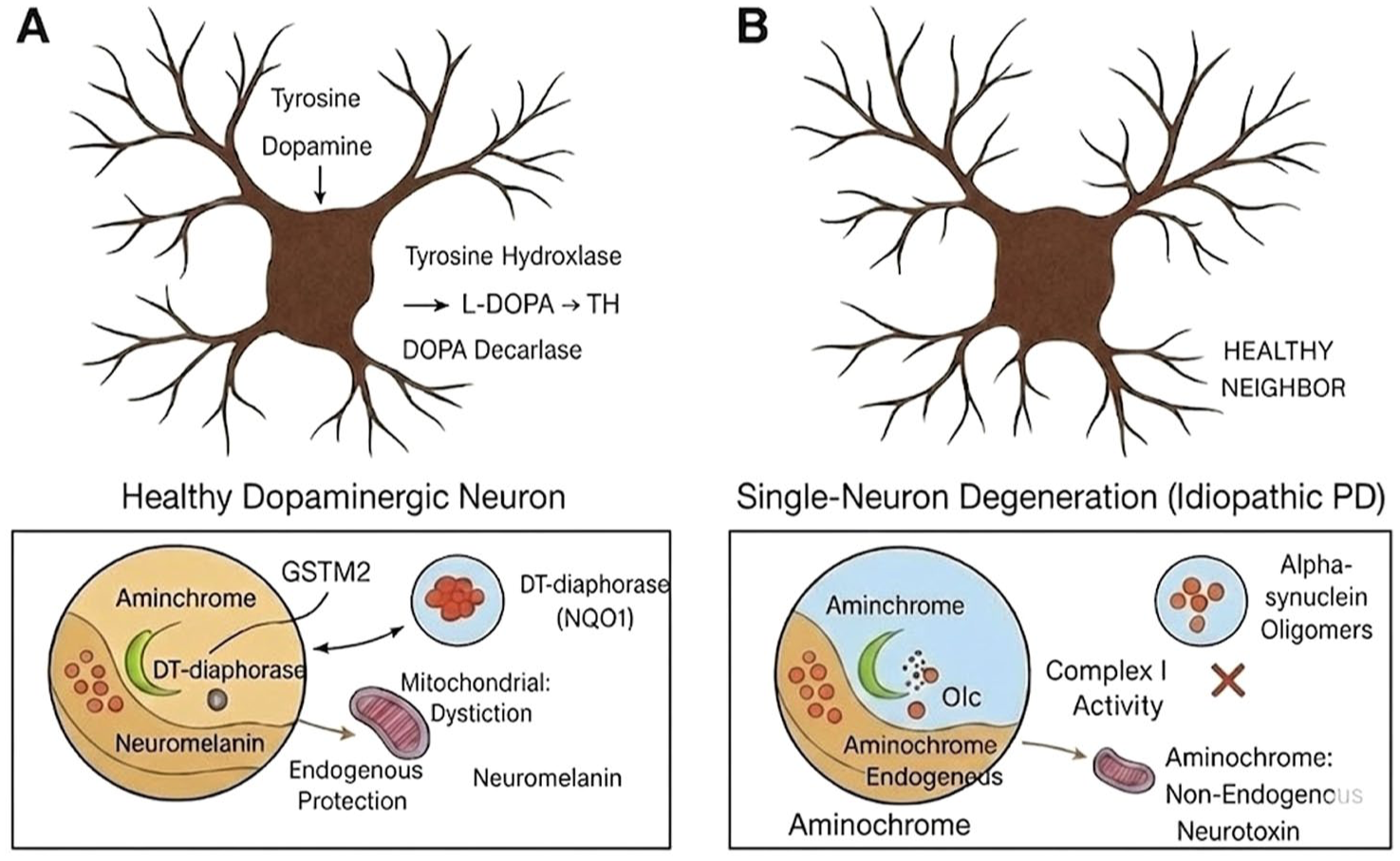
The single-neuron degeneration hypothesis in idiopathic Parkinson’s disease. In a healthy neuron (a), the neurotoxic intermediate aminochrome, produced during dopamine oxidation to neuromelanin, is neutralised by the protective enzymes DT-diaphorase and GSTM2, which is aided by the KEAP1/NRF2 pathway to prevent neurotoxicity. In single-neuron degeneration (b), the neutralisation capacity is overwhelmed. Aminochrome accumulates, inducing mitochondrial dysfunction, oxidative stress and the formation of toxic alpha-synuclein oligomers. Crucially, aminochrome is non-expansive and affects only that individual neuron, explaining the extremely slow loss of 58–72 neurons per day seen in iPD. The accompanying figure has been substantially revised to eliminate typographical errors (including ‘Aminchrome’, ‘Mitochondrial Dystiction’, ‘DOPA Decarlase’ and the incorrect label ‘Aminochrome: Non-Endogenous Neurotoxin’) and to clarify the schematic logic. Aminochrome is correctly labelled as an endogenous neurotoxin throughout.
Flaws in clinical trials and preclinical modelling
The validity of the single-neuron hypothesis is reinforced by the failure of recent clinical trials. Two Phase II clinical trials for monoclonal antibodies against alpha-synuclein aggregates, cinpanemab and prasinezumab, were stopped after failing to demonstrate positive outcomes.
Inappropriate preclinical models
A major contributor to the translational gap specifically observed in the PASADENA and SPARK trials is the reliance on toxin-induced models (like MPTP) or viral-vector overexpression models to justify immunotherapy. 90 Reference 21 in the present manuscript (Feany and Bender) refers to a Drosophila model of PD, not a mouse model; this distinction is noted, and the text is corrected accordingly. These models induce rapid, synchronised neurodegeneration over days or weeks, artificially inflating the apparent role of extracellular spreading. Consequently, success in clearing extracellular alpha-synuclein in these acute models falsely predicted efficacy for prasinezumab and cinpanemab. In reality, human iPD involves the slow, asynchronous and continuous endogenous metabolic erosion of individual neurons. Evidence for this slow progression derives from detailed morphometric post-mortem studies demonstrating the gradual, region-specific loss of neuromelanin-containing dopaminergic neurons across decades.24,91 If the pathology does not spread in massive, synchronised waves as seen in these preclinical models, then capturing extracellular alpha-synuclein with monoclonal antibodies provides little to no neuroprotective value.
The mathematics of neuron loss
The slow progression of PD presents a statistical challenge for clinical trials evaluating extracellular-targeted therapies. Detailed morphometric post-mortem studies – including those by Fearnley and Lees and Cheng et al. – estimate that a patient loses approximately 58 to 72 neuromelanin-containing dopaminergic neurons daily during the symptomatic phase.24,91 Even if prasinezumab or cinpanemab were partially effective at halting hypothetical cell-to-cell spread, they would at best prevent the loss of a fraction of these ~60 neurons per day. The MDS-UPDRS, which served as the primary endpoint in both the PASADENA and SPARK trials, is completely insensitive to such microscopic preservation over a standard 52-week trial period. The natural fluctuations in symptomatic scores and the placebo effect easily mask the preservation of a few thousand neurons, making it mathematically improbable for these specific antibody trials to demonstrate a statistically significant clinical benefit.92–94
Patient heterogeneity and misdiagnosis
The entity we clinically label ‘Parkinson’s disease’ is likely a syndrome rather than a singular disease. Clinicopathological studies suggest a misdiagnosis rate of up to 25% in early-stage disease. 94 Patients enrolled in trials may have Multiple System Atrophy (MSA), Progressive Supranuclear Palsy (PSP), or vascular parkinsonism. Antibodies targeting alpha-synuclein will almost certainly fail in patients whose parkinsonism is driven by tauopathy or vascular ischaemia. Even within ‘true’ PD, there are distinct molecular subtypes, such as GBA-PD, LRRK2-PD and iPD. Critically, wild-type alpha-synuclein forms oligomers in all these subtypes; familial mutations modulate the rate and characteristics of aggregation but are not the sole source of oligomeric toxicity. A ‘one-size-fits-all’ antibody approach dilutes the potential signal if the therapy is only relevant for a specific subgroup, masking potential efficacy.
The late intervention problem
The most critical reason for the failure of cinpanemab and prasinezumab is the fundamental mismatch between the timing of the intervention and the spatial location of the toxicity. By the time patients were enrolled in these Phase II trials, the neurodegenerative process had likely been active for 10 to 20 years. 95 According to the Single-Neuron Degeneration Hypothesis, surviving neurons are already burdened with severe intracellular autotoxicity – specifically aminochrome accumulation, mitochondrial collapse and internal alpha-synuclein oligomerisation. 75 Because full-sized monoclonal antibodies like prasinezumab and cinpanemab cannot penetrate the cell membrane, they are restricted to clearing extracellular debris. Removing extracellular alpha-synuclein offers no survival advantage to a neuron whose internal metabolic machinery is already failing. Figure 2 outlines the multifactorial reasons behind the lack of success in PD clinical trials.
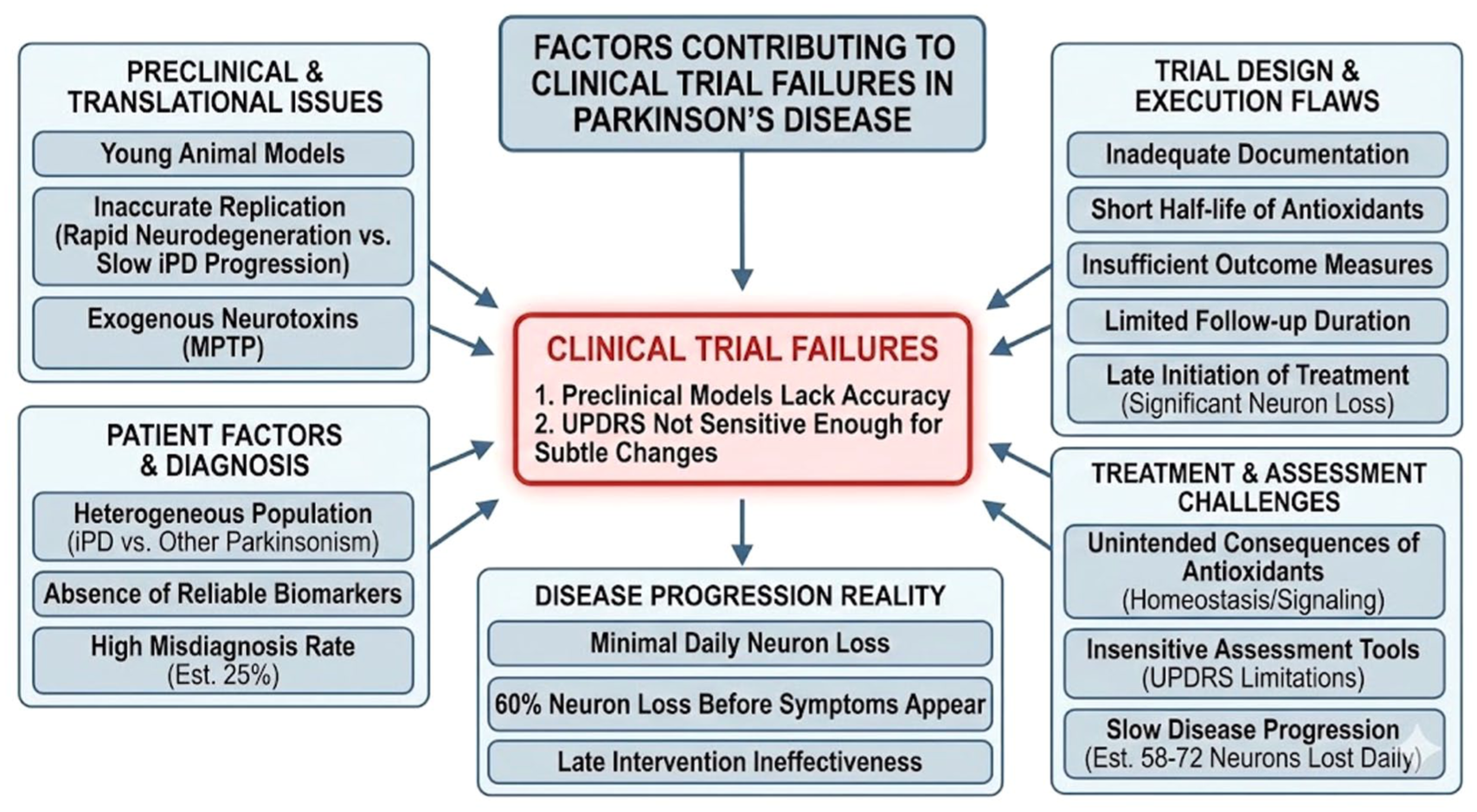
Factors contributing to clinical trial failures in Parkinson’s disease.
Blood–brain barrier paradox
Therapeutic development faces a complex BBB paradox. In early-stage PD, the BBB is largely intact, severely limiting the entry of peripherally administered antibodies (typically less than 0.1% cross into the CNS). Conversely, while BBB permeability may increase in late-stage disease due to neuroinflammation, the neurodegenerative burden is likely too advanced for the antibody to be effective. This ‘timing vs access’ conflict necessitates the development of receptor-mediated transport shuttles to enhance delivery during the early, treatable windows of the disease. Relevant strategies under investigation include engineering bispecific antibodies that exploit transferrin receptor–mediated transcytosis and the use of liposomal encapsulation targeting BBB receptors. Additionally, tissue distribution, cellular trafficking and the ultimate fate of antibodies following transcytosis are important pharmacokinetic determinants of CNS efficacy that must be optimised in next-generation designs.
Recommendations for future clinical trials in Parkinson’s disease
To overcome the challenges that led to the failure of recent clinical trials, future studies must be fundamentally redesigned to account for the disease’s extremely slow, gradual degenerative process.89–93,97 If the single-neuron degeneration hypothesis is correct, trials aimed at developing disease-modifying drugs should implement three primary strategic shifts.
First: Refined patient selection and biomarker-driven stratification
Patient selection must be refined by strictly enrolling only patients diagnosed with iPD, excluding atypical parkinsonism. A primary recommendation is the implementation of biological stratification to ensure that participants possess the specific molecular target the therapy intends to modify. Rather than relying solely on clinical symptoms, trials should utilise seed amplification assays (SAAs) to confirm the presence of pathological alpha-synuclein aggregates and distinguish between various conformational strains of the protein, 98 as different conformational folds may exhibit varying degrees of resistance to specific antibodies.99,100 SAAs are recommended here as a patient stratification tool to confirm bona fide iPD, not as an endorsement of extracellular alpha-synuclein as the therapeutic target. Furthermore, there is a critical need to intervene upstream in the disease’s progression. Intervening in the prodromal phase – before extensive dopaminergic neurodegeneration has occurred – offers the best opportunity for disease modification, as the ‘failed’ antibodies may simply have been administered too late to rescue damaged circuits.
Second: Enhanced outcome measures and combination strategies
Researchers should integrate objective digital health technologies and wearable sensor platforms to capture subtle, real-time changes in motor and non-motor function that traditional rating scales miss. 92 The adoption of validated digital endpoints could substantially reduce the measurement noise inherent in clinical assessments. Trials must prioritise imaging methods – such as SPECT (Single-Photon Emission Computed Tomography) – to accurately track the loss of neuromelanin-containing dopaminergic neurons at study initiation and completion. Given that PD involves oxidative stress, mitochondrial dysfunction, neuroinflammation and protein aggregation, a multi-pronged approach targeting several pathways simultaneously may be more effective than monotherapy. Finally, the pharmacokinetic profile of antibodies must be optimised to ensure better CNS penetration, addressing tissue distribution, cellular trafficking and antibody fate following transcytosis, which are all critical determinants of CNS efficacy. Nanobody- and micro-antibody–based approaches – owing to their reduced molecular size and favourable pharmacokinetic properties, including better tissue penetration and potential for intracellular access – may overcome some of the limitations associated with full-length monoclonal antibodies and merit prioritisation in the next wave of therapeutic development.
Third: Novel preclinical models targeting intracellular pathways
A concerted effort is required to develop preclinical models that better simulate the slow degenerative process of iPD. Since current technology makes proof-of-concept testing at the single-neuron level impractical, the most viable approach is to test whether a new drug can activate the KEAP1/NRF2 signalling pathway.101,102 Activation of this pathway is critical because it boosts the expression of protective enzymes – namely DT-diaphorase and glutathione transferase – essential for shielding neuromelanin-rich dopaminergic neurons from aminochrome-induced neurotoxicity. This directly addresses the gap exposed by the antibody trial failures. Since antibodies failed to clear extracellular seeds sufficiently to halt disease, or because toxicity is primarily intracellular, the KEAP1/NRF2 pathway offers a mechanism to armour the neuron from the inside against autotoxicity that antibodies cannot reach.
NRF2 (nuclear factor erythroid 2-related factor 2) is the master transcriptional regulator of the cell’s antioxidant and detoxifying response. Under basal conditions, NRF2 is sequestered in the cytoplasm by its repressor KEAP1. Upon oxidative stress or electrophilic challenge, KEAP1 undergoes conformational change, releasing NRF2, which translocates to the nucleus and binds the Antioxidant Response Element (ARE), initiating transcription of numerous cytoprotective genes, including DT-diaphorase (NQO1) and GSTM2. 102 Activation of this pathway is specifically relevant to aminochrome neurotoxicity: increased DT-diaphorase neutralises aminochrome within dopaminergic neurons, while astrocyte-derived GSTM2 – transferred via exosomes – provides additional intercellular protection. By bolstering these inherent defence systems, the cell can resist the slow, single-neuron degeneration proposed in iPD. Table 1 summarises the traditional versus proposed trial framework.
The traditional framework versus proposed model based on the single-neuron degeneration hypothesis.
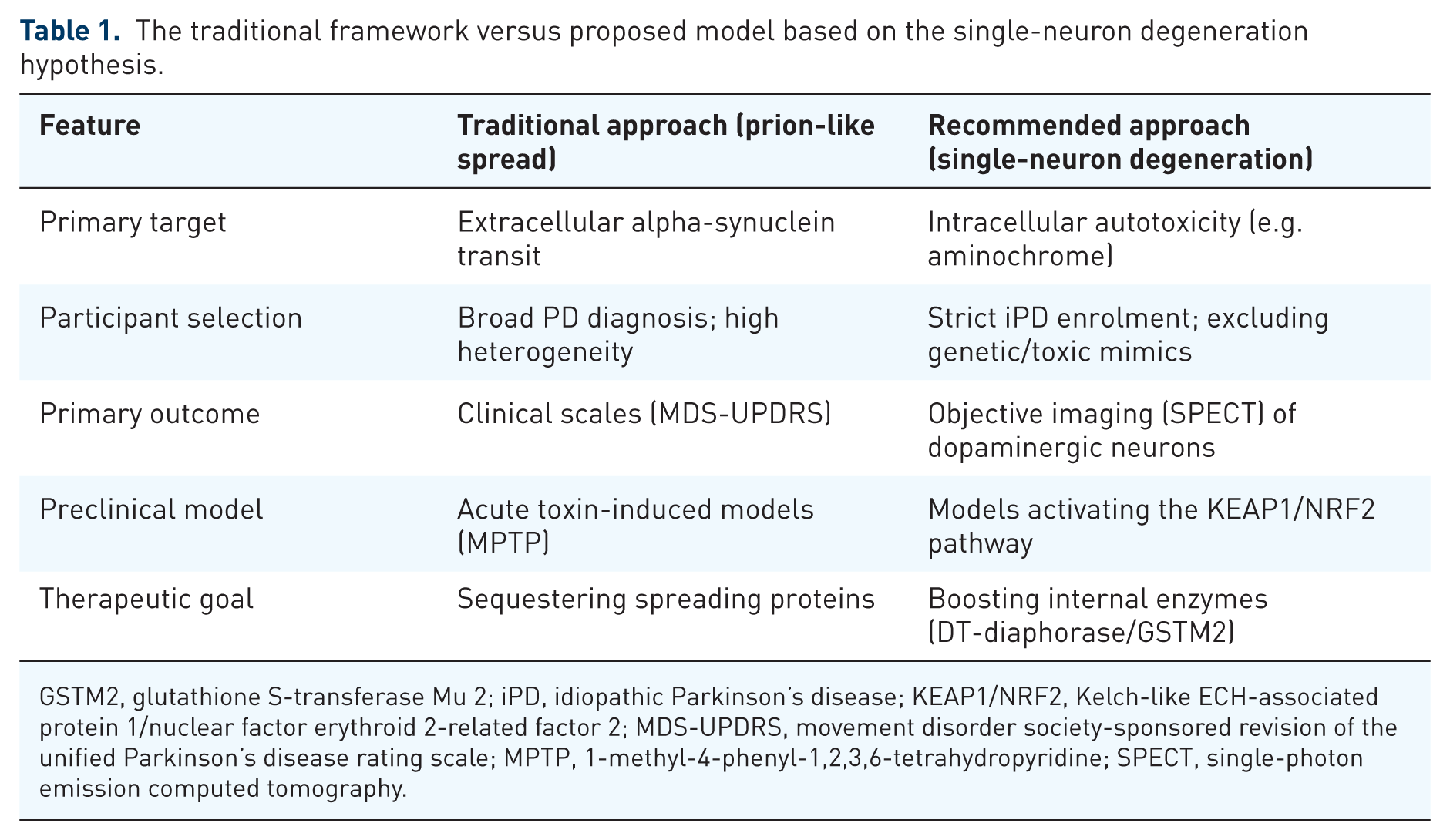
GSTM2, glutathione S-transferase Mu 2; iPD, idiopathic Parkinson’s disease; KEAP1/NRF2, Kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2; MDS-UPDRS, movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; SPECT, single-photon emission computed tomography.
Conclusion
The critical role of neurotoxic alpha-synuclein oligomers in the degeneration of dopaminergic neurons is well-established, making the strategy of blocking their harmful effects with monoclonal antibodies appear initially promising. However, this therapeutic approach has failed in clinical trials due to a confluence of factors. Primarily, there is a fundamental mismatch between the extracellular action of full-sized antibodies and the predominantly intracellular nature of autotoxicity (aminochrome accumulation, ER stress, mitochondrial collapse). Furthermore, preclinical justification relied heavily on acute neurotoxin models that trigger rapid, widespread neurodegeneration, fundamentally contradicting the slow, asynchronous progression of human iPD.
Detailed morphometric post-mortem studies estimate that neurodegeneration in idiopathic PD proceeds at a rate of approximately 58 to 73 neurons lost per day.24,91 Because of this extremely subtle and continuous decline, clinical measures like the MDS-UPDRS are unlikely to detect neuroprotective effects from monoclonal antibodies over a standard trial period.
These conclusions do not, however, imply that immunotherapy should be abandoned. The Braak staging model and the extensive literature on alpha-synuclein propagation provide compelling evidence for extracellular spread as a contributor to disease progression. Ongoing innovations – including engineered BBB-crossing antibodies, targeted protein degradation strategies, nanobodies and micro-antibodies – represent promising next steps that address the pharmacokinetic limitations of first-generation agents.
Future clinical trials seeking to slow the progression of idiopathic PD must undergo a paradigm shift. If the single-neuron degeneration hypothesis is correct, trials must: actively eliminate clinical heterogeneity through objective biomarkers (such as SAAs); prioritise imaging endpoints (such as SPECT) over subjective clinical scales; intervene in the prodromal phase; and develop drugs that activate the KEAP1/NRF2 signalling pathway – effectively armouring the neuron from the inside by boosting DT-diaphorase and GSTM2 expression. A combined strategy targeting both extracellular alpha-synuclein propagation and intracellular metabolic vulnerabilities represents the most promising path forward.