
Abstract
Synucleinopathies are a group of neurodegenerative disorders characterized pathologically by the presence of neuronal and/or oligodendrocyte inclusions composed of aggregated alpha-synuclein (αS). These disorders can be divided into two major sub-types: Lewy body disease and multiple system atrophy, which have distinct clinical and pathological profiles. As is seen with other neurodegenerative diseases, recent evidences suggest that the conformation and molecular organization of αS amyloid filaments deposited in brains might dictate the distinct pathological and clinical profiles in synucleinopathies. Resolving the structure of disease-specific αS aggregates becomes, therefore, of clinical interest. Until recently, the specific conformations of these aggregates at molecular resolution have remained elusive. This is now possible due to the advent and development of cryo-electron microscopy (cryo-EM) technology. This review aims to cover the recent advances in the structural biology of αS amyloid filaments implicated in synucleinopathies, raising a number of critical questions that continue to be investigated. What can we learn from these structures? What are the main structural differences between experimental systems and human tissue derived protein aggregates? What factors drive these differences? Do the amplified assemblies represent faithfully the seed structures? How these new findings improve our understanding of the molecular mechanisms behind fibril formation in disease? Can this knowledge be exploited for the design of diagnostic and therapeutical strategies? These aspects and the recent advances in the field will be addressed and discussed in this work.
Plain language summary
Filling the gap between the structure of alpha-synuclein aggregates and disease in Parkinson and related neurodegenerative disorders
Neurodegenerative diseases, such as Parkinson's disease, are associated with the buildup in the brain of a protein named alpha-synuclein. Instead of remaining in its normal form, this protein can aggregate and form structures that interfere with the normal function of nerve cells. These diseases, known as synucleinopathies, include Parkinson's disease, dementia with Lewy bodies, and multiple system atrophy, which differ in their symptoms and progression. The synucleinopathies involve the same protein but it is still not fully understood why each type of disease differ clinically and pathologically from one another.
In this context, recent advances in techniques such as cryo-electron microscopy make it possible to decipher the structures of the alpha-synuclein aggregates extracted from human brains. The studies revealed clear structural differences between alpha-synuclein aggregates generated in different synucleinopathies. Furthermore, recent evidences indicate that aggregates produced in the laboratory fail to match the structures of brain-derived aggregates, suggesting that the cellular machinery play a key role in guiding the conformational organization of these aggregates in human brains.
The scope of this review is to cover the main advances in the field of structural biology of synucleinopathies. The role of potential factors driving the different structures in a cellular context is discussed, how these findings contribute to a better understanding of the molecular mechanism behind disease, as well as how this knowledge may impact in the development of new diagnostic tools and most effective therapies are addressed in this work.
Introduction
Under normal conditions, the amino acid sequence of a protein defines its three-dimensional structure, which determines its biological function. In contrast, in neurodegenerative pathologies, the protein filaments deposited in post-mortem human brains indicate that the same protein can adopt many different amyloid structures.1–9 More and more evidence support the fact that the conformation and molecular organization of amyloid aggregates might play a key role in the pathological and clinical profiles of the different diseases.4,10 In that context, amyloid structures themselves might influence directly the disease, as different structures can affect their cellular propagation and toxicity in the brain, causing dysfunction and loss of brain cells. Consequently, the amyloid filament fold provides a basis for studying the molecular pathogenesis of these diseases, and at the same time for developing new therapies and diagnostic tools based on knowledge of these structures.
Determining the specific structures of aggregates derived from human brains at molecular resolution is now possible due to the technological development of cryo-electron microscopy (cryo-EM).11–17 This technique allows for the study of high molecular weight proteins or protein complexes that have been impossible by other methods, maintains the native state of a sample in solution, and can capture more than one conformation of a protein in the same experiment. Indeed, the development in cryo-EM is the main reason behind the steep rise in the number of deposited structures of brain-extracted amyloid filaments from a variety of neurodegenerative diseases. However, it is also true that amyloid fibrils derived from autopsied human brain tissue are a scarce resource. For that reason, laboratory-made aggregates from recombinant protein constitute the main tool for modeling the structure and biochemistry of neurodegenerative aggregates.8,9 Although these aggregates are relatively easy to fabricate, laboratory-derived structures have so far not accurately represented the structures of amyloid filaments deposited in human tissues.10,18–21
The above reveals key differences between experimental systems and human tissue-derived protein aggregates, indicating that the cellular environment of different brain cells could guide the formation of disease-specific folds.10,22 Identifying those cellular factors that drive the formation of different structures may therefore provide insights into the molecular mechanisms of pathogenesis, improving disease modeling in the laboratory.
The scope of this review covers the recent advances in the structural biology of synucleinopathies,23–25 a group of neurodegenerative disorders characterized pathologically by the presence of neuronal and/or oligodendrocyte inclusions composed of aggregated alpha-synuclein (αS). What we can learn from these structures, how these new findings improve our understanding of the molecular mechanisms behind fibril formation in disease, and how realistic it is that this knowledge can be exploited for the design of diagnostic and therapeutical strategies are some of the critical aspects that will be addressed and discussed in this work.
Synuclein-associated amyloid diseases
Synucleinopathies
It was in 1817 that James Parkinson published a short monograph, “An Essay on Shaking Palsy”, describing patients with tremors and slow movements. 26 However, it was Jean-Martin Charcot who at the end of 1800 proposed to call the pathology as Parkinson disease (PD), including the rigidity factor. Later, in 1912, Friedrich H. Lewy reported the presence of intracellular inclusion bodies in different regions of the brains of PD patients. A few years later, in 1919, Konstantin Tretiakoff identified neuronal loss and neuronal inclusions in the substantia nigra of patients, and use the term “Lewy bodies” to name these structures.27–31 Lewy bodies are now considered a pathological hallmark of PD, Parkinson Disease with dementia (PDD) and Dementia with Lewy Bodies (DLB).25,30,31 In 1969, Graham and Oppenheimer used the term “multisystem atrophy” to describe disorders characterized by glial cytoplasmic inclusions (GCI), also known as Papp-Lantos bodies.32–34 Initiating the 1990 decade a non-amyloid component in senile plaques (NACP) was identified by Tsunao Saitoh, 35 that resulted to be a synaptic protein identical to αS. 36 In 1997, Polymeropoulos discovered a mutation in the gene for αS (SNCA) in Southern Italian families of Greek heritage with PD. 37 In parallel, Spillantini reported that αS was the major component of Lewy bodies in sporadic PD and DLB patients. 38 Overall, these findings established the disease concept of “synucleinopathy”. Finally, it was reported that the major protein component in GCI was also αS, linking multiple system atrophy (MSA) and Lewy body disease (LBD). 39
Synucleinopathies are clinically and pathologically heterogeneous diseases characterized by neuronal or oligodendrocyte inclusions, or both, composed of aggregated αS in the form of Lewy bodies, Lewy neurites, and glial cytoplasmic inclusions.24,25 Pathologically, synucleinopathies can be divided into two major sub-types: LBD and MSA.34,40–48 Lewy body diseases consist of PD, PPD and DLB, that are considered part of the same spectrum of diseases, with clinical and pathological similarities.40–45 On the other hand, MSA is clinically and pathologically distinct from PD, PPD and DLB.34,39,42,46,47
In order to understand how the same protein can be associated with different pathologies, preliminary studies using αS aggregates derived from MSA (GCI-αS) and LBD (LB-αS) diseases were conducted to analyze their conformational properties. Proteinase K (PK) digestion profiles showed mostly undigested αS for GCI-αS, whereas LB-αS was digested into smaller fragments. 48 The structural differences between GCI-αS and LB-αS were further confirmed by immunostaining disease brain sections with a monoclonal antibody specific for a synthetic αS strain.48,49 The next step was to determine whether the structural variations observed between GCI-αS and LB-αS impacted on their seeding activities. Contrasting with spontaneous aggregation, seeded aggregation is based in the usage of brain-derived filament preparations as templates to seed in vitro filament formation by recombinantly purified monomeric proteins. 41 In that direction, extracts from MSA post-mortem brains were shown to induce aggregation of αS in cultured cells expressing wt and A53 T human αS. 50 By contrast, αS isolated from LBD disease showed no effect. 51 Consistent with the highly aggressive nature of MSA, brain-derived αS fibrils from GCI were shown to have a more potent seeding activity (∼1000 times) in vitro and in vivo than that derived from LBD disease, with MSA fibrils being also more potent inducers of motor deficits, neurodegeneration, and αS pathology.48,52 Indeed, αS preformed fibrils (PFF-αS) that were passed through oligodendrocytes were also more potent than those using other cell types, indicating that the GCI-αS strain is generated in an oligodendrocyte cell context. 48 Then, by incubation of αS monomers with cell lysates from primary oligodendrocytes and neurons, it was demonstrated that the production of GCI-αS strain depends on specific ‘factors’ in oligodendrocytes. 48 Overall, these preliminary works showed that oligodendrocytes, but not neurons, converted misfolded αS into a GCI-like strain, emphasizing that distinct αS strains are mediated by different intracellular milieus. 48
The protein alpha-synuclein
Alpha-synuclein (αS) is an intrinsically disordered protein encoded by the αS (SNCA) gene.53–55 It is predominantly located in the presynaptic nerve terminals of the central nervous system, where it regulates neurotransmitter release and synaptic function.40,56
Structurally, the protein contains 140 amino acids distributed in three distinct regions (Figure 1): the amphipathic N-terminal (residues 1–60), with KTK motifs involved in lipid binding,57–60 the highly hydrophobic central region (residues 61–95), required for the assembly of αS into amyloid filaments,61,62 and the acidic C-terminal region (residues 96–140), critical for blocking rapid assembly of αS into filaments.63,64 Interestingly, most of the familial SNCA gene mutations are localized in the N-terminal region, including: G14R, 65 V15A, 66 A30P, 67 A30G, 68 E46K, 69 H50Q, 70 G51D, 71 A53E, 72 A53V, 73 A53T 37 and K58N 74 (Figure 1).
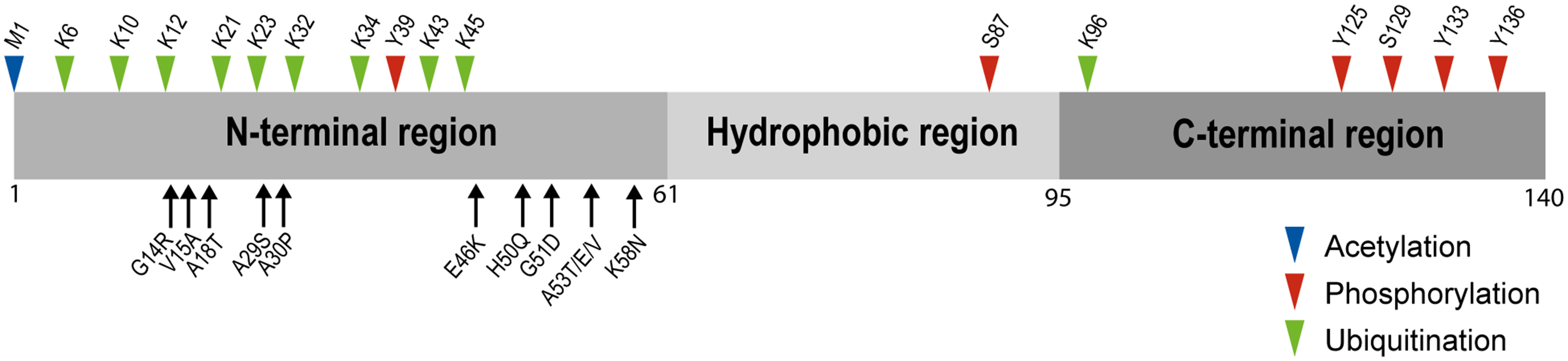
Structural features of αS. The full-length protein αS, showing the N-terminal, hydrophobic and C-terminal regions. Missense mutation sites at the N-terminal region associated to familial Parkinson's disease are shown (below the regions diagram). Post-translational modifications such as N-terminal acetylation (methionine), ubiquitination (lysines) and phosphorylation (serines and tyrosines) are labeled (above the regions diagram).
Under physiological conditions, the protein molecules adopt an ensemble of disordered conformations within the cytoplasm and/or undergoes a disorder-to-helix transition upon binding to membranes.75–77 However, under pathological conditions, the monomeric protein misfolds and follows a nucleation-dependent aggregation model, in which αS molecules self-associate, undergo conformational changes, and finally adopt the cross-β structure that is typically found in the αS amyloid protofilaments (Figure 2). 78 Subsequently, individual protofilaments can intertwine and bind with one another through a hydrophobic steric zipper interface to form larger filaments, adopting helical structures of different symmetry and pitch (Figure 2). 79
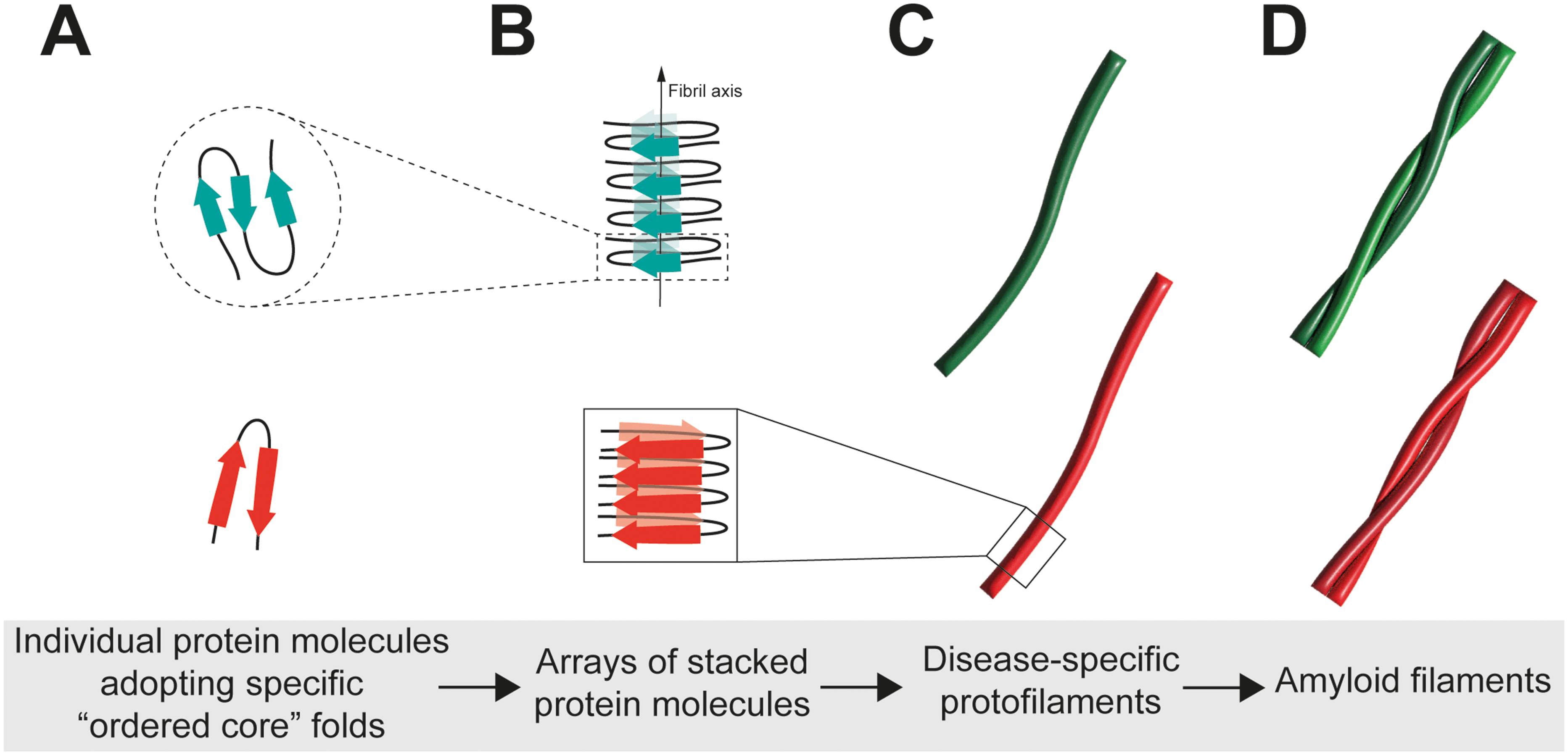
Ultrastructural organization of αS amyloid fibrils. (A–C) Individual protofilaments are made of stacked copies of αS molecules in identical conformations, forming the typical cross-β structure. The conformation of the αS molecules in a single protofilament is called the protofilament fold (i.e., Lewy fold, MSA fold, JOS fold). Specific folds are associated with different sub-types of synucleinopathies. (D) Individual protofilaments can intertwine and bind with one another to form larger filaments adopting different helical structures.
Recent works have revealed that αS aggregation kinetics and the structure of the resulting filaments are modulated by specific sequence motifs in the N-terminal region of αS monomers, flanking the hydrophobic central region, where the sequence 36GVLYVGS42 was shown to play a key role.80,81 Deletion of this motif prevented αS aggregation in vitro and also suppressed aggregation-induced toxicity in vivo. Based on the sensitivity of αS aggregation to pH and ionic strength, it has been suggested that this “master regulatory sequence” controls αS aggregation by a delicate balance between hydrophobicity and charge, that involves a network of local and distal intra- and intermolecular contacts.80–83
In terms of molecular organization, the αS monomers that form part of such amyloid structures adopt a variety of unique folds and then stack together to form the array of protein-specific protofilaments. These monomeric folds do not involve the entire protein but instead comprise a smaller ordered core, while the rest of the protein backbone remains unstructured and is part of what is termed as the ‘‘fuzzy coat’’.8,9 In most cases, the cores of αS filaments extracted from the brains of patients or assembled from recombinant protein in vitro encompass around 70 amino acids that extend approximately from residue 30 to residue 100, with half of the N-terminus (aa 1–30) and almost the entire C-terminal region forming the fuzzy region.5–9 As will be discussed later, each ordered core may adopt multiple structural organizations (polymorphs),5–9 whereas emerging evidence indicate that the specific conformation of each αS polymorph could also define a different disease type.5–7
In general, the formation of amyloid filaments has been understood as a mechanism to arrest aggregation by converting the unstable and more-toxic structures into stabilized and less-toxic forms. However, it is now known that the mature filaments are inherently unstable: they fragment into protofilaments of different sizes (seeds), which then amplify amyloid formation beyond the levels of spontaneous aggregation, with the concomitant effect that the newly formed amyloids adopt the same structure as the seeds. 84 Indeed, as in other aggregopathies, αS filaments have been suggested as the species responsible for the transmission and progression of the pathology. In that scenario, aberrant protein filaments can be propagated through the brain during disease progression via a prion-like mechanism. 85 When pathogenic filaments are introduced into a native cell, they can template the folding of endogenous proteins into the same aberrant conformation, resulting in fibril growth and formation of amyloid structures. Thus, identification of disease-associated core folds can shed light on the mechanisms behind cellular propagation and explain the structural basis of disease severity and progression.
Structural organization of αS amyloid fibrils
Alpha-synuclein filaments derived from human-brains
The hypothesis that the clinicopathologic heterogeneity of synucleinopathies is linked to different strains of αS is supported by recent cryo-EM-based structural studies demonstrating that αS filaments derived from brains affected by PD, PPD and DLB share a unique specific structure known as the “Lewy fold”, 5 whereas native αS filaments in MSA possess an amyloid structure that is markedly different from the Lewy fold. 6 These findings revealed, as for the tau protein, 4 the existence of distinct molecular conformers of αS filaments that are associated with different synucleinopathies. More recently, a different amyloid fold was reported in a case of juvenile-onset synucleinopathy (JOS),7,86 whereas a Lewy-MSA hybrid fold was observed in cases of atypical forms of MSA. 87
Regarding the structural features of the αS filament folds reported to date, studies focused on MSA showed that the MSA fold has two types of filaments – type I and II – that are made each of two different protofilaments (A and B) (Figure 3B and Table 1).6,10 Both types of filaments are asymmetrical and have a left-handed helical twist. The ordered core in the different protofilaments of the MSA fold extends from G14 to F94 (for 1A and 2A), K21 to Q99 (for 1B), and G36 to Q99 (for 2B). The protofilament cores consist of an extended N-terminal arm and a tightly packed C-terminal body. In all cases, the conformation of the core sequence is stabilized by a characteristic salt-bridge between residues E46 and K80. 6
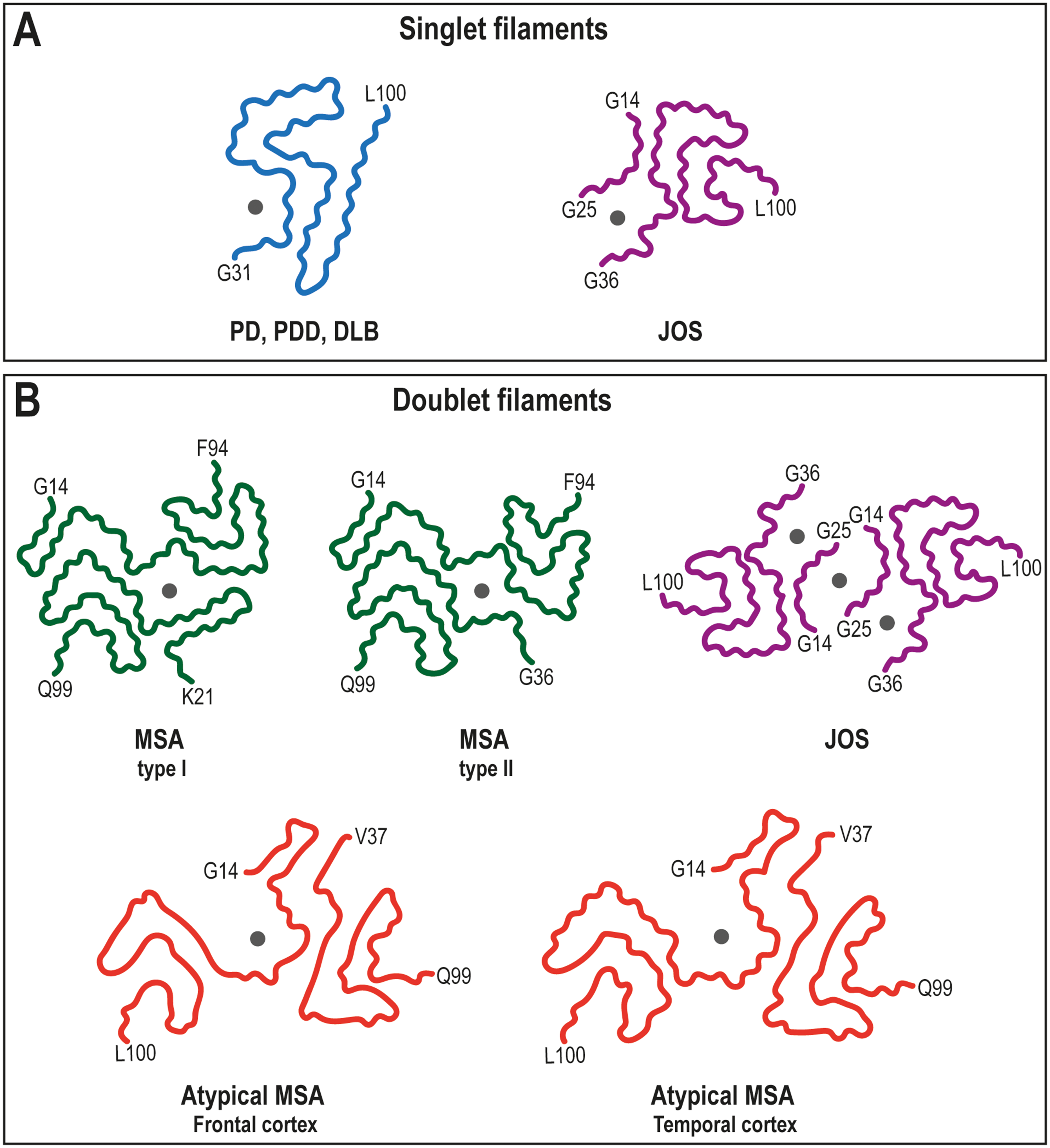
Structures of disease-relevant amyloid folds of αS fibrils in synucleinopathies. Main-chain traces of the αS protein fibrils with positions of unknown densities (grey dots). Structural organization of αS as singlet or doublet filaments and the disease diagnosis associated with each filament fold is indicated. First and last residues of the ordered core are indicated for each structure. PDB IDs of the structures that are shown: In panel A, 8A9L (singlet filament – Lewy fold – from PD, PDD, DLB) and 8BQV (single filament from JOS). In panel B, 6XYO (doublet filament from type I MSA), 6XYP (doublet filament from type II MSA), 8BQW (doublet filament from JOS), 9OBP (doublet filament from atypical MSA, frontal cortex) and 9E9X (doublet filament from atypical MSA, temporal cortex).
Structure of predominant polymorphs found in vitro and ex vivo αS fibrils.
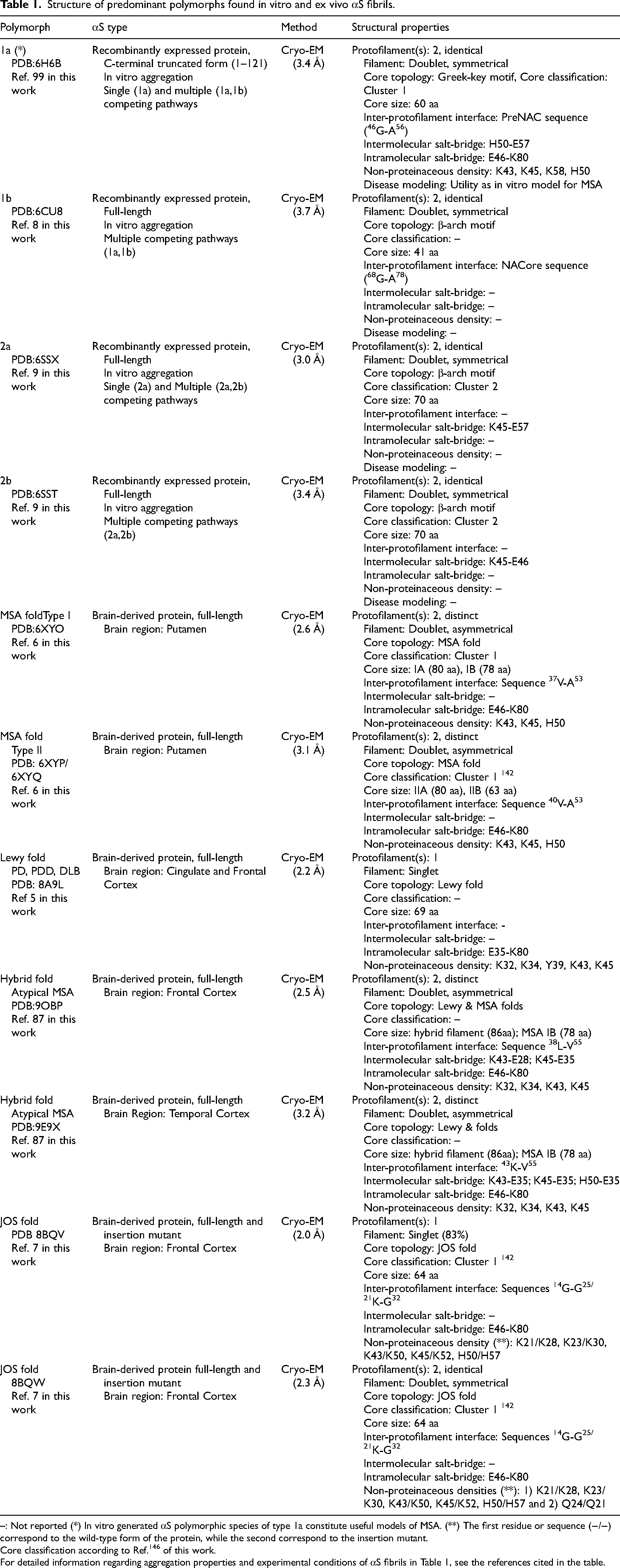
–: Not reported (*) In vitro generated αS polymorphic species of type 1a constitute useful models of MSA. (**) The first residue or sequence (−/−) correspond to the wild-type form of the protein, while the second correspond to the insertion mutant.
Core classification according to Ref. 146 of this work.
For detailed information regarding aggregation properties and experimental conditions of αS fibrils in Table 1, see the references cited in the table.
The inter-protofilament interface contains the V37-A53 and V40-A53 sequences for the type I and type II filaments, respectively. Interestingly, for both types of MSA filaments, non-proteinaceous densities are present at the interface of the two protofilaments, in a cavity surrounded by the side-chains of residues K43, K45, and H50 from each protofilament (Figure 3B and Table 1). These extra densities are not connected to the protein density and its chemical nature remains to be established. There are also unconnected densities next to external lysines (i.e., K58 and K60) that have not been identified. Similar peripheral densities were observed in tau structures and may be negatively charged molecules of the fuzzy coat.1,88–90
Compared with the doublets seen in MSA, the cryo-EM structures of αS filaments from the brains of individuals with PD, PDD and DLB are identical singlets. Their structures are made of a single protofilament arranged into the Lewy fold (Figure 3A and Table 1), 5 that is markedly different from the protofilaments fold of MSA. Structure determination of αS filaments with Lewy fold was difficult due to a lack of helical twist in many filaments. 5 In contrast to the left-handed species observed in MSA, those fibrils that did twist in LB disorders were characterized by right-handed helices. 6 In structural terms, the Lewy fold consist of an ordered core extending form residue 31 to 100, that arranges into a three-layered structure of β-strands, with a salt-bridge between E35-K80 contributing to stabilize the packing of the single protofilament. 5 A non-protein density is also observed in these filaments, located in a cavity formed by the side-chains of residues K32, K34, Y39, K43, and K45 (Figure 3A and Table 1). 5
Interestingly, αS filaments derived from the brain of a patient with juvenile-onset synucleinopathy (JOS) showed a distinct fold to those described for MSA and Lewy filaments (Figure 3A and B and Table 1).7,42,91,92 In JOS, the protein αS has 147 amino acids due to the insertion of the sequence MAAAEKT after threonine at position 22 of the sequence. Immunoblot analysis detected the presence of both wt αS (15 kDa) and the insertion mutant αS comprising 147 amino acids (16 kDa). 7 This is the first report of an insertion mutation in the SNCA gene. The majority of αS filaments (∼80%) from the patient with JOS comprises a single protofilament with an ordered core extending form residue 36 to 100, or 43 to 107 in the insertion αS mutant (Figure 3A). In the structure of the remaining αS filaments, doublets were observed, that are made of two identical, symmetrically packed protofilaments (Figure 3B). The conformation of the core sequence in both singlet and doublet JOS filaments is stabilized by a salt-bridge between residues E46 and K80, as described for MSA filaments. Both singlet and doublet filaments in JOS have a left-handed helical twist. The JOS fold differs from the Lewy fold, and is more similar to the MSA fold. The singlet JOS protofilament presents also a large non-proteinaceous density that is similar in size and environment to the unidentified density of Lewy fold (Figure 3A and Table 1). 7 In this case, an island formed by the sequences G14-G25 in wt αS or K21-G32 in the insertion αS mutant appears packed at the N-terminus of the ordered core, whereas the additional density surrounded by K21/K28, K23/K30, K43/K50, K45/K52, and H50/H57 appears located between this island and the main core (Figure 3A and Table 1). On the other hand, in the case of JOS doublet filaments, the non-proteinaceous density is found mediating filaments interactions, as observed for MSA filaments (Figure 3B and Table 1). In this case, the density seems to be coordinated by neutral polar residues provided by two opposing islands (sequences G14-G25/ K21-G32) connected to each protofilament, 7 suggesting a different chemical nature that the large non-proteinaceous densities within singlet JOS filaments.
The current structure-based classification of synucleinopathies has been expanded by the recent finding that αS can adopt a Lewy-MSA hybrid fold in cases of atypical types of MSA. 87 Doublet filaments, in which two different protofilaments are asymmetrically packed against each other, were found in the cryo-EM structures of αS filaments obtained from neuronal inclusions in the temporal and frontal cortices of individuals with atypical MSA disease (Figure 3B and Table 1). 87 Substructures of the Lewy fold in the center and the MSA folds at the N- and C-terminus make up one protofilament (aa 14–100), which is referred to as a “Lewy-MSA hybrid protein”. The other protofilament holds a MSA fold (MSA-PF-IB). A cavity enclosing a non-proteinaceous strong density surrounded by the side chains of K32, K34, K43 and K45 is also found in the Lewy part of the hybrid filament (Figure 3B and Table 1). Overall, the doublet filaments structures from the temporal and frontal cortices are nearly identical, with variations at the level of the salt-bridges and hydrophobic interactions within their inter-protofilament interfaces. These changes render the doublet filament from the frontal cortex more rigid that the one form the temporal cortex. It's interesting to note that the Lewy-MSA/MSA doublet filament structure reported in that work constitutes the first example from human brains without any non-proteinaceous density at the inter-protofilament interface. 87
As mentioned in section 2.2, all αS filaments have a fuzzy coat that encompasses the amino- and carboxy-terminal regions of the protein. Besides the density for the ordered core of αS filaments, additional “fuzzy” densities (islands) are also detected in the Cryo-EM density maps. The reconstructed densities for these islands confirm that they are made of peptides corresponding to parts of the αS sequence that are N- and/or C-terminal of the ordered core, in which the connecting residues presumably adopt a disordered conformation. Such a peptide-like density, of unknown identity, is packed against residues K80–E83 of PF-IIA in MSA filaments form patient's brains. 6 In the case of the Lewy fold filaments, two additional islands appears packed against β5 and the N-terminal half of β9, respectively. 5 The reconstructed densities for both islands indicate that they are made of peptides, but a lack of distinct side chain densities precluded their identification. The JOS fold consists of a compact core, and two disconnected density islands, assigned to the 6KGLSKAKE13 and 14GVVAAAEKTKQG25 sequences in the 140-residue wild-type protein, and 21KTMAAAEKTKQG32 in the 147-residue insertion mutant protein. 7 The cryo-EM reconstructions of JOS doublet filaments show that the islands are essential for dimerization, with the islands from both protofilaments facing each other across the filament axis.
Recent evidences suggest that the interaction between the fuzzy coat and fibril core influences substantially the neuronal transmission of αS filaments. 93 The studies reveal that the spatial position between the dynamic fuzzy coat, especially the acidic C-terminal tails, and the structured core in the αS fibrils promote the formation of recurrent transient contacts between both regions. 93 Consequently, the effective molecular interface presented by pathological fibrils is defined not only by the ordered cross-β scaffold but also the fluctuating disordered segments that surrounds it. This scenario leads to the formation of different polymorphisms with distinct transmission capabilities, supporting a critical role of fibril's fuzzy coat in the seeding activity of αS filaments. 93 Altogether, these evidences reveal the critical roles of the αS fibril fuzzy coat in its pathological transmission process and suggests a novel strategy of intervention for such diseases. Because the fuzzy coats exist extensively in many pathological protein fibrils, and the biological function of the fuzzy coat within these pathological fibrils remains unknown, it would be very important to explore the functions of this knowledge gap in the future.
Disease-associated mutations in brain-derived αS filaments
Regarding the impact that familial mutations might have on the structures of ex vivo αS filaments, it has been reported that G51D and A53E variants can cause atypical, mixed MSA and PD pathologies.72,94,95 These residues are both located at the protofilament interface of the ex vivo MSA filaments, in close proximity to K43, K45 and H50. Structurally, these mutations may alter the steric and charge features around the central cavity, a fact that might lead to a change in the nature of the non-proteinaceous compounds within the central cavity and/or potentially affect the stability and pathogenicity of the mutated fibrils, triggering mixed MSA and PD pathology.
On the other hand, in JOS cases, cryo-EM modeling revealed that the insertion αS mutant could more readily adopt the ordered core structure than the wild-type species, although both species form part of disease samples. 7 This could help to understand how this mutation drives pathogenesis, as the mutant core could nucleate both wild-type and mutant proteins within the same cellular environment. In this case, disease-associated mutations can result in more labile core folds and a greater tendency toward fragmentation, which both contribute toward more efficient seeding.96,97 Indeed, prion-like proteins can seed their own aggregation and propagation through the brain, but some strains nucleate more readily than others or seed aggregates at different rates. 98
Overall, the structure of fibrils from patients with familial mutations will need to be solved in order to confirm the predicted effects, to understand why those mutations specifically cause disease and to enhance our knowledge about the development and progression of sporadic disease as well.
Recombinant αS fibrils generated in the laboratory
In recent years, cryo-EM has yielded several structures of in vitro assembled filaments of recombinant αS.8,9,97,99–113 The first studies reported the presence of two predominant polymorphs, that were termed “rod” (1a) 99 and “twister” (1b), 8 (Figure 4A and B and Table 1). Both polymorphs were composed of two identical protofilaments, that are symmetrically packed. The ordered core for polymorphs 1a and 1b comprises 60 and 41 residues, respectively. Although both the rod and twister polymorphs contain a bent-β arch structure (comprising the region between residues H50 and V77), the additional ordering of residues at the C terminal in the rod polymorph results in the formation of a β-strand-rich structure with a Greek key-like topology. The residues forming the inter-protofilament contacts are another difference between the rod and twister polymorphic species. In the rod polymorph, the hydrophobic interface between opposing protofilaments is formed by the preNAC region of the protein (46GGVVHGVTTVA56), which is stabilized by the hydrophobic contacts between the side chains of A53 and V55. 99 In addition, H50 and E57 at the edge of the steric zipper core, together with K45, form electrostatic interactions that stabilize further the inter-protofilament interface. There are another two pairs of electrostatic interactions, i.e., K58-E61 and E46-K80, that form intramolecular salt-bridges, important for the folding and packing of the Greek-key topology. In contrast, the interface in the twister polymorph contains the NACore region of the sequence (68GAVVTGVTAVA78) 8 (Figure 4B and Table 1). In conclusion, the rod and twister filaments have structurally conserved kernels that come into contact at either the preNAC or NACore sequences. As a result, the location of the protofilament packing interface, rather than the kernel structures, is the factor that determines the fibril polymorphism.8,99
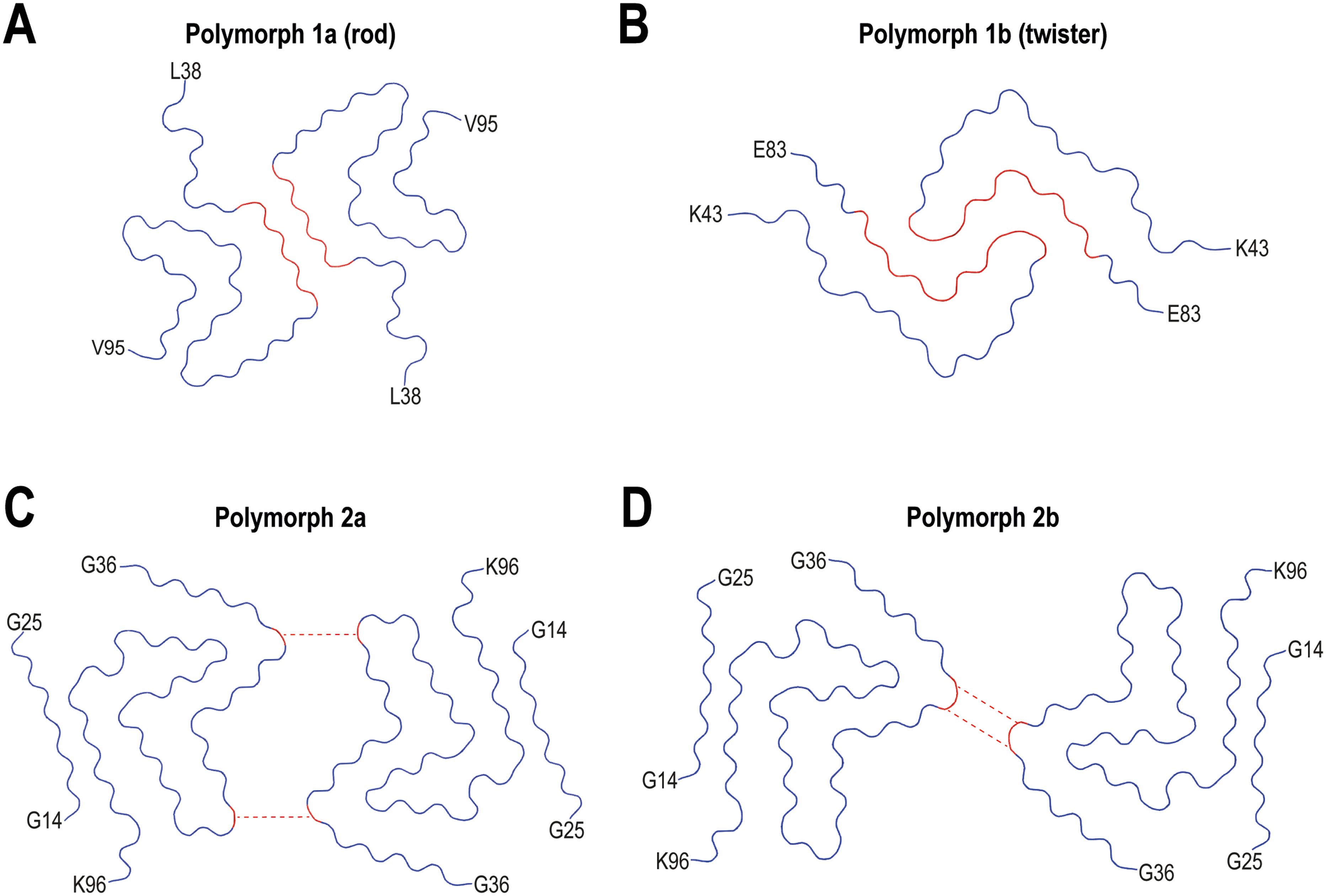
Structures of recombinant αS fibrils assembled in vitro. Cryo-EM structures of the rod (A) and twister (B) polymorphs of wt αS are shown, as well as 2a (C) and 2b (D) polymorphs. The ordered core structures of each fibril are shown. Inter-protofilament salt-bridges are represented by dotted lines. First and last residues of the ordered core are indicated for each structure. PDB IDs of the structures that are shown: 6H6B (Rod polymorph, 1a), 6CU8 (Twister polymorph, 1b), 6SSX (polymorph 2a) and 6SST (polymorph 2b).
Subsequent research revealed the formation of two novel polymorphs, named αS polymorph 2a and αS polymorph 2b, to distinguish them from the previously described αS fibrils 1a and 1b (Figure 4C and D and Table 1). 9 The 2a and 2b polymorphs are formed by two symmetrically packed protofilaments. In both cases, the polymorphs 2a and 2b exhibit bent β-arch motifs, with ordered cores made up of 70 residues. In contrast to the hydrophobic interaction between protofilaments in polymorph 1a and in 1b, the interface in the polymorphs 2a and 2b is stabilized by electrostatic interactions through salt-bridges between residues K45 and E57, or K45 and E46, respectively.
Regarding the origin of the distinct polymorphs all fold one structures (polymorphs 1a and 1b) were grown under buffer conditions with either a polyanion or a large chaotropic negative ion,8,99 whereas polymorphs 2a and 2b were grown under phosphate-free conditions with the only anion being Cl−. 9 Added to that, the aggregation conditions used in each study (time, temperature, stirring) were also different.
Interestingly, a highly positive clustering of K43, K45, K58, and H50 residues is observed at the surface of polymorph 1a, next to the salt-bridge H50-E57. This configuration requests the anchoring of a counter-ion to neutralize the repulsion of the three positively charged residues, as it is supported by an observed density in the cryo-EM map.
Modeling disease-associated mutations in recombinant αS filaments
As previously mentioned, one important distinction between the in vitro αS structures is the type of protofilament-protofilament interaction. Several residues associated with familial Parkinson's disease (PD) are found in the dimeric interface of the rod polymorph (E46K, H50Q, G51D, A53E/A53T/A53V). In contrast, the distinct folds between polymorphs 1 and 2 place the familial PD mutation sites in a completely different environment. Considering the fold one structures, this indicates that PD-associated mutations may disrupt the hydrophobic interface between fibril cores in the rod polymorph, while having little impact on the stability of the twister polymorph, where the structure adopted by the core pushes the mutation sites at periphery away from the interface. Structurally, the G51D and A53T/E mutations introduce hydrophilic or even charged residues into the hydrophobic dimeric interface of the rod polymorph, which may cause the disruption of this structure, while the H50Q mutation may break the electrostatic interactions among H50, K45 and E57 from the opposing subunit. On the other hand, formation of the twister polymorph and the αS fibril polymorphs 2a and 2b are not directly affected by these mutations.
In polymorph 1a, the E46K PD familial mutation destabilizes the fibril structure by affecting the intramolecular salt-bridge between E46 and K80; however,105,112 in αS polymorph 2b this residue is part of the protofilament interface. Therefore, the charge repulsion between lysines K45 and K46 from the two protofilaments may interfere with the contacts between the two protofilaments in polymorph 2b. In fact, the mutant E46K adopts polymorph 2a structure, which results in lysines K45 and K46 from one subunit connecting with E57 in the other. Similarly, the K58N missense mutant promotes the formation of a salt-bridge between K45 and E57, which results in a protofilament interface formed by the residues K45-E57. 74
Interestingly, beyond the fact that the familial PD mutations are predicted to disrupt the wild-type fibril assembly, yet these mutants can still form amyloid fibrils. Indeed, the E46K mutant yielded different structures with and without N-terminal acetylation. The N-terminal acetylated E46K mutant αS fibrils showed a protofilament interface formed by the V74-Q79 residues, 105 whereas the non-acetylated variant showed an interface with K45-E57. The E46K mutant is toxic to neuronal cells. 112 These fibrils were less stable and easily fragmented, but were also a better seed than wild-type fibrils, suggesting that toxicity might be related to seeding rather than fibrils stability.105,107 While the mutation G51D yielded a structure with a fully disordered N-terminal region and an interface comprising residues V74, A76, and A78 (Figure 5A), 109 an interface composed of T59 and K60 residues was found in the N-terminal acetylated A53E mutant (Figure 5B). 113 Such a small interface was also found in the N-terminally acetylated A53 T mutant αS fibrils. 106 The H50Q mutation yielded two new polymorphs: narrow fibrils formed from a single protofilament and wide fibrils, composed of two protofilaments with T59-K60 at the interface (Figure 5C). 97 As mentioned above, the K58N mutant showed a protofilament interface formed by K45-E57, slightly shifted probably due to the proximity of the mutation site. 74
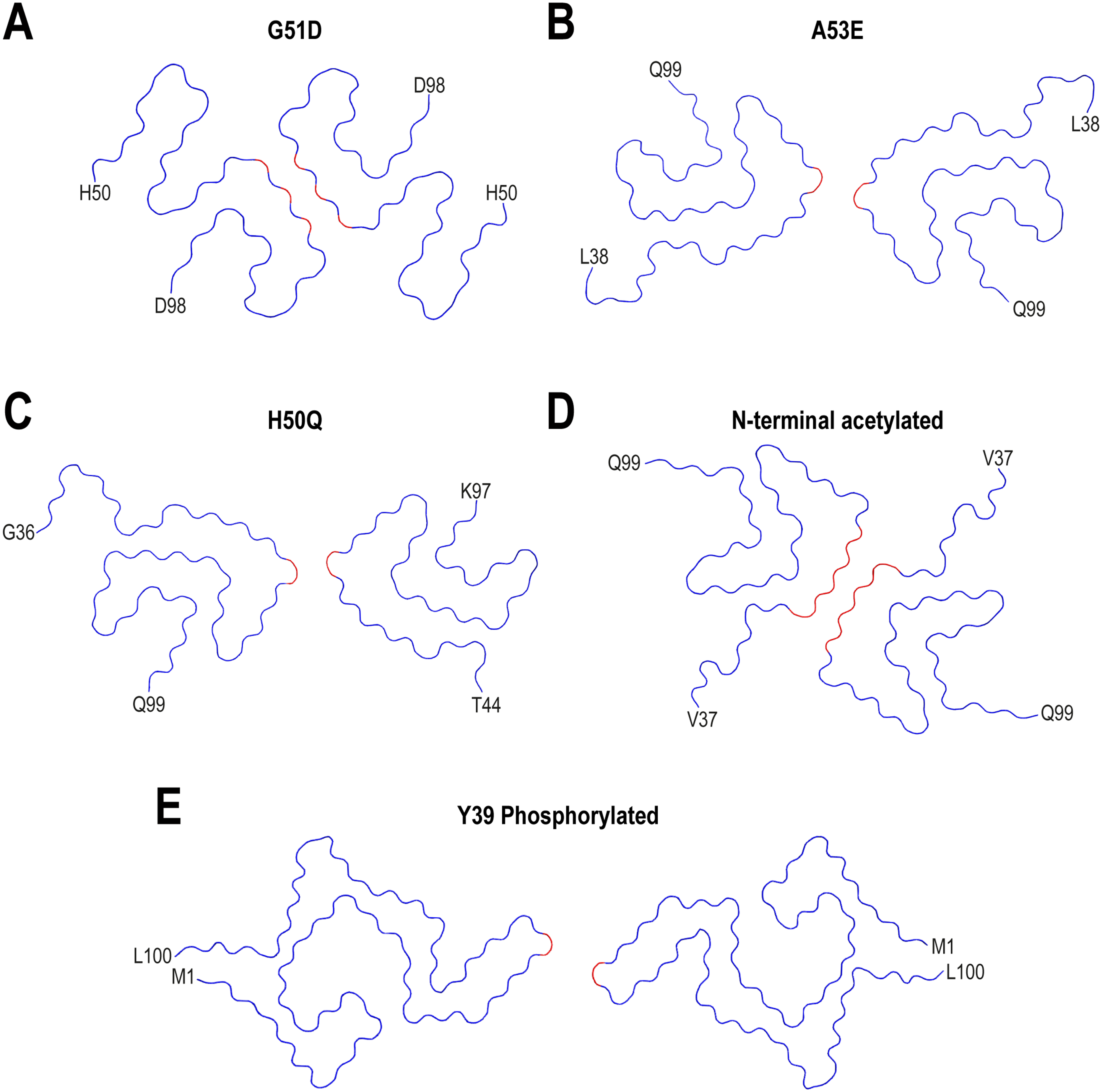
Impact of familial-mutations and post-translational modifications on the structure of recombinant αS fibrils assembled in vitro. Cryo-EM structures of G51D (A), A53E (B) and H50Q (C) mutants are shown. N-terminal acetylated (D) and Y39 phosphorylated (E) full-length αS fibril variants are also shown. The ordered core structures of each fibril are shown. First and last residues of the ordered core are indicated for each structure. PDB IDs of the structures that are shown: 7E0F (G51D), 7UAK (A53E), 6PES (H50Q), 6A6B (N-terminal acetylated) and 6L1T (Y39 phosphorylated).
Overall, these results demonstrate that missense mutations associated with familial PD can alter the conformation of protofilaments and their inter-protofilament interfaces, resulting in variable αS fibrils with distinct aggregation kinetics, seeding activity, stability and cytotoxicity.
What factor(s) drive the structural differences between ex vivo and in vitro αS fibrils?
In vitro fibrils have been extensively used as the default in vitro model for studying amyloid aggregates. However, the reported evidences indicate that both spontaneous aggregation of recombinant αS and seed amplification of fibrils extracted from brain produce αS filaments that fail to match the folds of aggregates that accumulate in human brains.20,100
As described in section 3.1, a common feature of all αS filament structures extracted from post-mortem patients is the presence of additional non-proteinaceous densities in the cryo-EM maps that cannot be attributed to αS. In an attempt to understand the origin of such a densities located at the periphery of fibril structures,6,18,89,114–117 within the inter-protofilament interface,6,7,114–117 and/or in internal pockets inside individual fibril cores,89,114–120 it has to be considered the involvement of filament-specific cofactors and/or post-translational modifications (PTMs) in the formation of the distinct structures.48,121,122 Furthermore, the broad range of structures observed in vitro contrasts with the structural homogeneity of amyloids within each disease, indicating that the cellular machinery may be crucial in directing the formation of specific amyloid structures in human brains.
Post-translational modifications
The importance of PTMs on protofilament folding has been shown in studies of the tau protein, where these modifications seem to dictate which fold the protein assumes, and each fold appears to be unique to each distinct tauopathy. 118
In the structures of brain-derived αS filaments, PTMs such as N-terminal acetylation, C-terminal truncation, ubiquitination and acetylation of lysine residues, as well phosphorylation of threonine, serine and tyrosine residues has been consistently found.6,10,123
The predominant presence of N-terminal acetylation and C-terminal truncation, ubiquitination of K6, K12, K21 and K23 residues, phosphorylation of S129, Y133 and Y136, deamination of Q62, N65, N103, Q109 and N122, dimethylation of K80 and trimethylation of K60 were detected in the sarkosyl-insoluble fraction (αS filaments) from MSA and LBD patient's brains.6,124 In addition, in some cases, the αS filaments from MSA patient's brains showed ubiquitination of K60 and K80, acetylation of lysines contained in the region 12–102 (K12, K23, K32, K34, K45, K58, K60, K80, K97, K102), methylation of K12, K60, K80 and K96, oxidation of M1, M5, M116, M127 and H50, as well as phosphorylation of Y39, T59, T64, T72, T81 and T92.6,124 Overall, these findings indicate that a combination of PTMs rather than single modification might influence the structure and stability of αS filaments in synucleinopathies.
In agreement with these findings, disease-specific PK-resistant profiles found in LBD and MSA were attributed to different C-terminal truncation events,48,125,126 while immunoblot analysis of sarkosyl-insoluble αS from LBD and MSA brains revealed the presence of distinct high-molecular αS bands, consistent with a disease specific PTM profile.127,128 Indeed, in LBD cases, the processing at the C-terminus of αS occurs at the sides of D119 and N122 residues, while in MSA cases the processing occurred at multiple sites between the 114–123 regions. 124 Furthermore, differences in PTMs in the core region between DLB and MSA, especially the acetylation level of lysine residues, may be significant. 124
Finally, in αS filaments from JOS, acetylation of residues K34, K45, K58, and K60, methylation of K43, K58 and K96, oxidation of M1 and H50, and phosphorylation of T33 and Y39 were detected, in addition to PTMs that are relatively common in MSA and LBD. 124 The PTMs pattern of JOS was similar to that of MSA, but very different from that of LBD.
Although more research is required to determine the PTM profiles of synuclein diseases in more detail, as well as their contributions to the architectures adopted by ex vivo filaments, an structural analysis revealed that a relationship could be established between the nature of PTMs and the type of protofilaments that is formed in ex vivo MSA fibrils. 6 Indeed, it was predicted that type II large protofilaments in MSA would be impacted by phosphorylation at T72 and ubiquitination at position K80. 6 Based on the protofilament structures, K80 ubiquitination is only compatible with protofilament IIA, since it would conflict with the surrounding densities in the other protofilaments. Interestingly, ubiquitination of K80 was detected in sarkosyl-insoluble α-synuclein in MSA cases showing preponderance of type II filaments. 6 Similar to ubiquitination, phosphorylation may also affect fibril type. The T72 residue may be favored for phosphorylation in protofilament IIA over IA in the MSA fibrils because IA has T72 buried, while IIA has T72 exposed to a cavity. 6 Additionally, because the side chain of T81 is solvent-exposed in PF-IIB2 but buried in PF-IIB1, phosphorylation of this residue may distinguish between PF-IIB1 and PF-IIB2. 6
On the other hand, research on in vitro-generated αS fibrils mostly concentrated on the structural impacts of N-terminal acetylation, C-terminal truncation and phosphorylation. Previously, studies focused on the impact of PTMs on the aggregation propensity of αS demonstrated that phosphorylation at S129 and Y39 promotes aggregation, while these modifications at S87, and Y125, Y133 and Y136 residues inhibit aggregation. 129 Similarly to the effects induced by glycosylation of T72 and S87 residues, oxidation of methionines at positions 1, 5, 116 and/or 127 of the protein sequences was shown to inhibit aggregation. 129 On the other side, nitration of tyrsones located at the N- and C-terminal region, in all cases, promoted aggregation. 129 Finally, truncation of C-terminus was shown to accelerate aggregation, while N-terminal acetylation reduced the oligomerization of the protein. 129
With regard to the structural studies, even if in vitro filaments do not fully recapitulate ex vivo structures, these works demonstrated that PTMs can affect the packing of specific regions and the overall folding of protofilaments. As shown in Figure 5D, N-terminal acetylation of full-length αS yielded a variant with identical structure to polymorph 1a. As mentioned previously, the E46K mutation yielded different structures with and without N-terminal acetylation.105,107 For the non-acetylated E46K variant, the inter-protofilament interface comprised the segment K45-E57, whereas it was formed by the V74-Q79 residues for the N-acetylated variant.
Regarding C-terminal truncation, these truncated species showed an increased seeding activity compared to full-length αS.64,130–135 C-terminal truncation of different lengths (1–103, 1–122) in combination with N-acetylation yielded fibrils where the hydrophobic interface formed by the segment H50-E57 is maintained (Figure 4A). 103 Cryo-EM structures of these species showed a tighter packing of their filaments cores compared to longer αS, a fact that together with the better propensity to seed aggregation might account for filament formation in vivo. On the other hand, N-terminal truncated construct resulted in the formation of a structure with two asymmetric protofilaments composed of a bent β-arch kernel (E46-K96) and an extended β-hairpin (E61-D98), where the inter-protofilament interface comprised the segment K80-E83. 136
Finally, studies focused on the phosphorylation of Y39 and S129 residues indicated that pS129 yielded identical structures to polymorph 2a, 9 with pY39 resulting in the formation of twist-dimer and twist-trimer fibrils with interface comprising E57-K58 (Figure 5E) 104 The formation of an electrostatic interaction network at the N-terminal region involving the phosphorylated Y39 tyrosine and eight positively charged lysines, together with the occurrence of changes at the filament core structure, support the potential role of PTMs in the modulation of αS fibril conformation. 104
Molecular cofactors
The noncovalent binding of molecular cofactors contributed by the different cellular environments and their potential role in the formation of the specific αS filament structures found in synucleinopathies became an active area of research in the last years. 137 In a seminal work, to test whether the generation of GCI-αS strain relies on cell structures or specific ‘factors’, αS monomers were incubated in the presence of cell lysates from primary oligodendrocytes and neurons in which cell structures were disrupted but cell ‘factors’ were preserved. 48 The results showed that the generation of GCI-αS strain depends on specific ‘factors’ in oligodendrocytes. Additionally, it was demonstrated that, regardless of the presence of the protein in the extracts, spinal cord homogenates from donor mice may cause the formation of amyloid structures and disease phenotypes in an animal model of αS. 138 These findings support the idea that substances present in cells play a role in how αS folds into fibrils, affecting αS filament formation and seeding. As mentioned before, in the case of MSA, the non-proteinaceous is located at the binding interface between the two protofilaments, in a cavity formed by the side-chains of residues K43, K45, and H50. 6 In the case of Lewy fold structures, the non-proteinaceous density appears located at a surface cavity formed by residues K32, K34, Y39, K43, and K45. 5 The extra densities found at the periphery of the αS filaments in the JOS and hybrid Lewy-MSA fold are of a similar nature to that reported in Lewy and MSA folds.10,87 While lysine is the primary coordinating residue at these cavities, and paired lysines are a common motif in tau and αS fibrils, 137 other basic (arginine and histidine) and polar neutral residues (asparagine, glutamine, serine, threonine and tyrosine) may also participate, as seen in MSA and LBD filaments. 6 In all cases, the molecular cofactor binding at these locations may act to stabilize the rather highly positive clustering made of lysines, allowing αS fibrils to preserve their structure. It is interesting to note that the local concentration of cofactors may also influence the protofilament-protofilament packing in mature filaments where these binding cavities exist between two protofilaments, such as in MSA fibrils. This may result in more labile or stable core folds with a different tendency to fragmentation, which contribute to modulate the seeding activity of the filament. This might help also to understand why in vitro fibrils default to thermodynamically stable but non-physiological folds.
To date, although their polyanionic nature is suspected, the non-proteinaceous molecules have not been identified and the mechanism by which they influence αS fibril polymorphism in synucleinopathies is unknown. Considering the naturally charged properties of αS, various charged biopolymers have been proposed as potential candidates to interact with αS filaments within the cellular context. Due to the varied structures and properties of polyanions, they may bind to positively charged residues located at different sites of αS, co-assembling with the filaments to form distinct structural polymorphs. Regarding identity of molecular cofactors, when combined with its inferred polymeric and anionic nature, candidates such as RNA, heparan sulphate and acidic polypeptides emerge. In a recent study exploring heparin-induced assembly, extra densities attributed to negatively-charged heparin were observed in the proximities of residues K43-K45 and K58-K60. 139 The study demonstrated that heparin not only modulated the fibrillar assembly but also modified the fibril structure over time, leading to the formation of multiple polymorphs.139,140 Moreover, heparin-like oligosaccharides could also block neuronal uptake and propagation of formed αS fibrils, suggesting a pharmaceutical potential for these heparin derivatives. Studies using computational chemistry hypothesized that the non-proteinaceous density in αS fibrils in post-mortem brain tissue from PD, PDD, and MSA may be poly(ADP-ribose) (PAR), a negatively charged polymer generated by PAR polymerase-1. 141 Added to previous works that demonstrated that PAR binds to αS and may accelerate its fibrillization, 142 the study confirmed that PAR and αS complexation is driven by positively charged lysine residues in the protein. 141 Studies performed in the context of a lipidic system formed by POPC and POPA reported the formation of several polymorphic species: polymorph L1 had hydrophobic surfaces with adhered lipids, polymorph L2 resembled polymorph 2a, and polymorph L3 was similar to the structure corresponding to E46K. 101 Furthermore, polyphosphates may also be potential candidates, along with phosphatidylinositol phosphates and inositol phosphates, according to a recent study that used blind docking, molecular dynamic simulations, mutagenesis, and in vitro biochemical assays. 143 This is supported by a prior study that showed phosphatidylinositol-3,4,5-trisphosphate (PIP3) interacts with αS and starts its aggregation. 144
Further studies aimed at determining the identity of these cofactors are needed, as their local population could guide fibril formation and stabilization. In that context, the design of small molecules that can target cofactor binding sites could also be exploited therapeutically.
Open questions and future directions
The evidences discussed here highlights the complexity of the αS amyloid aggregation process and underscores the importance of developing laboratory conditions or experimental models that replicate brain-derived amyloid filamentous structures. The hypothesis that clinicopathologic diversity of synucleinopathies is associated to different strains of αS is supported by structural biology studies and seeding experiments. We now know the distinct amyloid folds that αS may adopt in synucleinopathies.5–7,10,87 The fact that distinct filament folds for αS characterize various forms of synucleinopathies, suggest that amyloid formations may play a role in disease. What remains unclear is how filaments relate to brain cell death in the different diseases. On this regard, the amyloid structures themselves may have a direct impact on disease, as distinct structures will have different biochemical and biophysical features that may affect their formation and spreading, their interactions with other cellular components and their toxicity in the brain. On the other hand, the mere existence of amyloid filaments, regardless of their forms, may cause brain cells to dysfunction and die. For example, aggregated fibril structures may also trap cellular cofactors, which could have detrimental effects on the cell and accelerate its degeneration. In both situations, structures of amyloid filaments are implicated and serve as a molecular foundation for studying the molecular mechanisms behind disease. Revealing what guides the formation of specific amyloid structures observed at the end stage of disease may help to understand better the cellular and subcellular origins of filament formation during the initial stages of pathology. It is possible that initial filament assembly takes place in a distinct cellular context, which may give rise to a specific structure. In that direction, new experimental cellular and animal models that may produce with high fidelity the specific disease folds will be crucial. 126
The potential factors driving polymorphism in ex vivo αS aggregates were also presented in this work. In that context, the molecular cofactors, PTMs and fuzzy coat hypotheses were discussed. The observation that the non-proteinaceous densities observed on the cryo-EM maps of αS amyloid structures tend to be disconnected from the protein densities favors the molecular cofactor hypothesis. With some exceptions, the percentage of αS molecules modified by PTMs at a given residue was shown to be low,6,10 which suggests further that PTMs might not be responsible for the additional densities in the cryo-EM maps of brain-derived αS filaments. However, the possibility of both effects occurring together cannot be ruled out. In terms of PTMs, the fact that the pathological structures observed in JOS are different from those in LBD and exhibit PTMs similar to those in MSA 124 suggests that, as reported for tau filaments, 145 the nature of the PTMs in αS filaments depends on the ordered core structure. In terms of fuzzy coat, due to technical limitations, it is not possible visualize directly the fuzzy coat structures as clearly as the rigid core regions in the αS fibrils, and we do not yet fully understand the exact interaction mechanisms between the fuzzy coat and core regions that result in the formation of different polymorphs, which is important and needs to be further explored in the future. Concomitant with these findings, the identity of the molecular cofactors should be known. Additionally, a greater number of samples needs to be evaluated, along with more disease cases, to establish these conclusions more firmly.
We now understand better the differences between ex vivo amyloid folds and laboratory amyloid folds. In a recent work, alignment and clustering of the fibril cores in most of published αS structures allowed classification of their tertiary structures into two major clusters: Cluster 1, characterized by a conserved Greek key motif spanning residues 69–95, and Cluster 2, characterized by a conserved β-arc motif spanning residues 51–67. 146 The work indicated that the conserved Geek key or β-arc motifs are likely formed during the early events of fibril assembly, becoming key targets for disease diagnosis and therapeutic intervention. In addition, the study concluded that Cluster 1 fibrils make attractive in vitro models of MSA. 146 Although this new structural framework might complement the disease-based classification, the majority of the structures analyzed by the authors came from in vitro preparations (∼65%), while ex vivo fibrils – those extracted from PD, PDD, DLB, MSA, and Juvenile Onset Synucleinopathy (JOS) patient tissue – represent the smallest proportion (∼8%).5–7,10 It is obvious that further work is required in this area.
In the framework of this review, an intriguing question in the field of neurodegenerative diseases is whether the reconstructed structures reflect accurately the in vivo conformations adopted by these amyloids and/or if the brain-derived filaments may be biased towards specific polymorphs. In fact, there are factors that may have a potential impact on the studied amyloid structures such as the methods used for extraction of amyloid filaments from brain tissues, the area of the disease brain from which the filaments were extracted and then, aspects that are unique to and inherent in the Cryo-EM technique. Indeed, there have been reports of the structural sensitivity of αS aggregates for various anatomical distributions of the protein in the brain. 6 For instance, of the 5 cases of MSA human-brain derived samples that were analyzed, type I filaments were predominantly found in the putamen in cases 1 and 3, while type II filaments were identified almost exclusively in the cerebellum in MSA case 1 and in the frontal cortex in MSA cases 2, 3 and 5. This clearly shows the influence that the region of isolation of αS filaments may have on their structural properties. When it comes to the limitations of the cryo-EM technique, it should be mentioned that the majority of filaments from PD, PDD and DLB cases did not show a helical twist in the cryo-EM micrographs. In fact, for each case studied, a minority of filaments, ∼25%, was twisted, which made it possible to determine their structure using helical reconstruction. However, the untwisted filaments structures were not solved. It is therefore impossible to rule out the possibility of additional, minority folds among untwisted filaments.
To complement the limitations mentioned in the previous paragraph, recent structural studies have shown the enormous potential of cryo-electron tomography (cryo-ET) to ascertain the in situ conformation and localization of amyloid fibrils in neurodegenerative diseases, 147 even at a much higher resolution. In situ cryo-ET investigations could serve as an alternative approach for Cryo-EM based studies and provide opportunities to assess the structural behavior of these amyloid filaments in a cellular context. In addition to Cryo-ET, Advanced Sensing of Aggregates–Parkinson's Disease (ASA–PD) was used recently to quantify αS aggregate density in disease-brain tissues, and determine how those species were distributed spatially and by size in post-mortem human brain tissue. 148 By addressing the regional and cellular microenvironments that promote the development of disease-specific species, as well as their temporal evolution, the ASA–PD platform can also be used to complement the Cryo-EM based studies. 148
According to a consistent message imparted in studies focused on the structures of αS amyloid filaments, fibrils become attractive targets for disease detection and therapeutic intervention. In fact, knowing the αS filament folds may be fundamental to develop tracers and biomarkers that can detect these fibrils in patients. Furthermore, the design of molecules that could disrupt the amyloid core folding would open the door to the development of structure-guided treatments. However, how realistic is it that disease-specific αS folds can be exploited for treatment and/or diagnosis? The invention and application of seed amplification assays (αS-SAA) in biological fluids and tissues has been a recent advance in biomarkers for diagnosis. Actually, the existing biochemical assays include protein misfolding cyclic amplification (PMCA) 149 and real-time quaking-induced conversion (RT-QuIC). 150 Although this biochemical assay has been clinically validated and is already offered commercially in some countries, its wider application is still restricted and limited due to the lack of laboratory standardization. Indeed, the biochemical profiles, structure, morphology, and phosphorylation pattern of human brain-derived αS fibrils from synucleinopathy patients differed from those of the “model” SAA fibrils when their biophysical properties were evaluated. 151 The fact that SAA fibrils failed to adopt the biochemical, structural, and PTM properties of the seed brain-derived αS fibrils, addresses a severe limitation of SAA in replicating faithfully the intrinsic biophysical properties of the seed fibrils. 151 These results underscore the need to reassess the SAA seeding technique and its ability to produce disease-relevant αS fibrils used in various in vitro and in vivo models. The models for templated seeding may also need reconsidering. Coupling the αS-SAA with single-cell collection methods will be also essential to investigate the contribution of the various brain regions to the production of αS seeds. In addition, the αS-SAA may be used in conjunction with other tests, such as the immunohistochemical analysis of skin biopsies 152 to fully validate the discriminatory ability of αS-SAA.
These observations illustrate clearly the limitations of using SAA fibrils to model brain-derived αS fibrils. This does not imply that tests involving seed amplification should always be avoided. They are helpful, for example, in discriminating between disorders like Parkinson's disease and MSA.153,154 However, the fact that the amyloid filament structures generated during amplification differ from those of the seeds must be taken into account when evaluating and interpreting them.
Regarding neuroimaging ligands, currently there are no validated imaging probes to detect αS pathology. Developing a robust imaging test to detect and visualize αS pathology in the brain is key to generate a biological definition of synucleinopathies. Future research should be directed to the search of additional markers of αS pathology and neurodegeneration to improve accuracy and sensitivity of detection.
With relation to the search of more effective therapeutic approaches, developing agents that can disrupt the folding of the amyloid core folding and interfere with their propagation and seeding profiles might aid in the formulation of a structure-guided therapies. In fact, designing small molecules that can target cofactor binding sites could be beneficial for the disaggregation of αS assemblies. To achieve this, the identity of the molecular cofactors should be known. The best strategy to determine this is by replicating in vitro and in biological models the structures of the amyloid fibrils obtained from the brain. Clearly, further work is needed in this area.
Footnotes
Acknowledgements
C.O.F. thanks Centro Universitario Argentino-Alemán (CUAA-DAHZ) for fellowship under the Binational PhD program “Molecular Bioscience and Biomedicine”. P.D.C.G. and I.F. thank CONICET for fellowship.
Funding
The author disclosed receipt of the following financial support for the research of this article: This work was supported by the Max Planck Institute for Multidisciplinary Sciences (MPINAT-MPG) [Code number P10390].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.