
Abstract
The lack of disease-modifying therapies for Parkinson’s disease (PD) places a severe burden on patients, their caregivers and aging societies. While the incomplete knowledge of underlying causes and pathophysiology remains a major bottleneck towards the development of rational therapies, there have been important advances in our understanding of neurodegenerative processes. Here, animal models have contributed substantially by allowing researchers to decipher PD-relevant pathology and the development of corresponding behavioral signs, which enabled development and refinement of treatment options. Without animal models, hypotheses of mechanisms underlying the development of motor symptoms after dopamine depletion, dyskinesia as side effects, the impact of diverse environmental neurotoxins, or the formation of alpha-synuclein pathology and spreading could not have been conclusively tested. Likewise, novel technologies are enabling the use of patient derived tissues for generating in vitro models that further enhance our understanding of mechanisms and of the effects of specific genetic modifications and their role in neurodegeneration. However, animal models are, as of yet, still required to model a fully functional central nervous system connected to the immune system and peripheral organs, which are impacted by the diversity of PD pathology and symptoms. Here, we provide an overview of key contributions of animal models, their ongoing role in PD research, and how this research can be enhanced by the concomitant use of advanced in vitro systems.
Plain language summary
Animal models and cell models to study Parkinson's disease
There is currently no treatment that can slow or stop the progression of Parkinson's disease (PD), which places a heavy burden on patients, caregivers, and aging societies. One major reason for this is that we still do not fully understand what causes PD. However, recent research has improved our understanding of the underlying disease processes. Animal models have played a crucial role in this progress. They allow scientists to study how PD-related loss of neurons lead to symptoms, and they have helped in developing and improving treatments. Without animal models, we would not be able to test important ideas about how dopamine loss leads to motor symptoms, why certain treatments cause side effects like dyskinesia, how environmental toxins contribute to the disease, or how abnormal proteins like alpha- synuclein spread in the brain. New models now let researchers study cells and tissues taken from patients. These tools make it easier to explore how specific genetic mutations contribute to brain cell damage. Still, animal models are currently the only way to study how Parkinson's affects the whole nervous system, including its connections to the immune system and other organs, all of which can be involved in the disease. This article reviews the important contributions of animal models to Parkinson's research, their ongoing value, and how combining them with advanced in vitro systems can improve our understanding of the disease and lead to better treatments.
Introduction: The growing burden of Parkinson’s disease
Parkinson's disease (PD) is the second most frequent progressive neurodegenerative disorder. 1 According to the World Health Organization there are about 8.5 million individuals suffering from PD in 2019. 2 Worryingly, PD is the fastest growing neurological disease in aging societies, and the global prevalence is expected to further double within the next 20 years (up to 2% in people over the age of 60 and 6% in people over 80 years). 2 PD is defined as a motor syndrome with dopamine neuron loss in the substantia nigra pars compacta, accompanied by aggregation of the protein alpha-synuclein (aSyn) in Lewy bodies.3,4 Since neurodegeneration and protein accumulation can only be detected postmortem, the clinical diagnosis relies on characteristic motor symptoms of Parkinsonism that include bradykinesia, resting tremor, rigidity, and postural instability. 4 These symptoms are mainly caused by the progressive dopaminergic neurodegeneration and consequently, lack of the movement controlling neurotransmitter dopamine. Alterations in non-dopaminergic neurotransmitter systems add to the complexity of the disease and are associated with non-motor symptoms.5–8 Importantly, these symptoms, such as olfactory deficits, rapid-eye-movement sleep behavior disorders, and autonomic dysfunction, precede the overt motor deficits by years or even decades. 9 While their occurrence may indicate an increased risk to develop PD, they do not lead to reliable risk assessments and are not validated as endpoints for clinical trials.
The available dopamine replacement therapy efficiently reduces the motor symptoms, but does not slow down disease progression, does not treat most non-motor symptoms and is limited in dose and duration due to the development of L-DOPA induced dyskinesia. There is currently no therapy available to prevent, halt or cure this debilitating disorder, which places a very high burden on patients and caregivers. At time of diagnosis patients are still highly engaged and active members of our society, part of the work force and central to the well-being of their families. The economic burden of PD solely for the US has been estimated at around $51.9 billion for 2017, including direct and indirect medical costs, reduced employment, disability income, and reduced loss in volunteer work. 10 In Europe, the total costs for PD alone in 2010 amounted to €13.9 billion. 3
These facts underline that significant efforts are necessary in order to develop disease-modifying treatments. However, this requires further research into the disease pathomechanisms, discovery of novel drug targets, and drug development towards clinical trials. To achieve this, researchers need to conduct basic science studies, using a variety of model organisms and systems. While enormous efforts have been focused on replacing in vivo experiments in animals by using ex vivo and in vitro systems, the fact is that there is still no alternative to animal models for establishing drug efficacy and safety prior to clinical trials in human subjects. The urgency to develop treatment provides the ethical backbone for the use of animal models, strictly following the 3R principles, which are now part of good laboratory and scientific practice in research. There have been several reviews providing in-depth overviews of animal and cellular models of PD, including by our groups. Therefore, here we discuss key findings (summarized in Table 1) that were learned from using animal models to highlight their essential role in PD research and how they enabled numerous benefits for patients. Furthermore, we discuss complementary ex vivo and in vitro approaches to highlight their potential as well as their current limitations to reduce, refine or replace in vivo experiments.
Key learnings from commonly used animal models of PD.
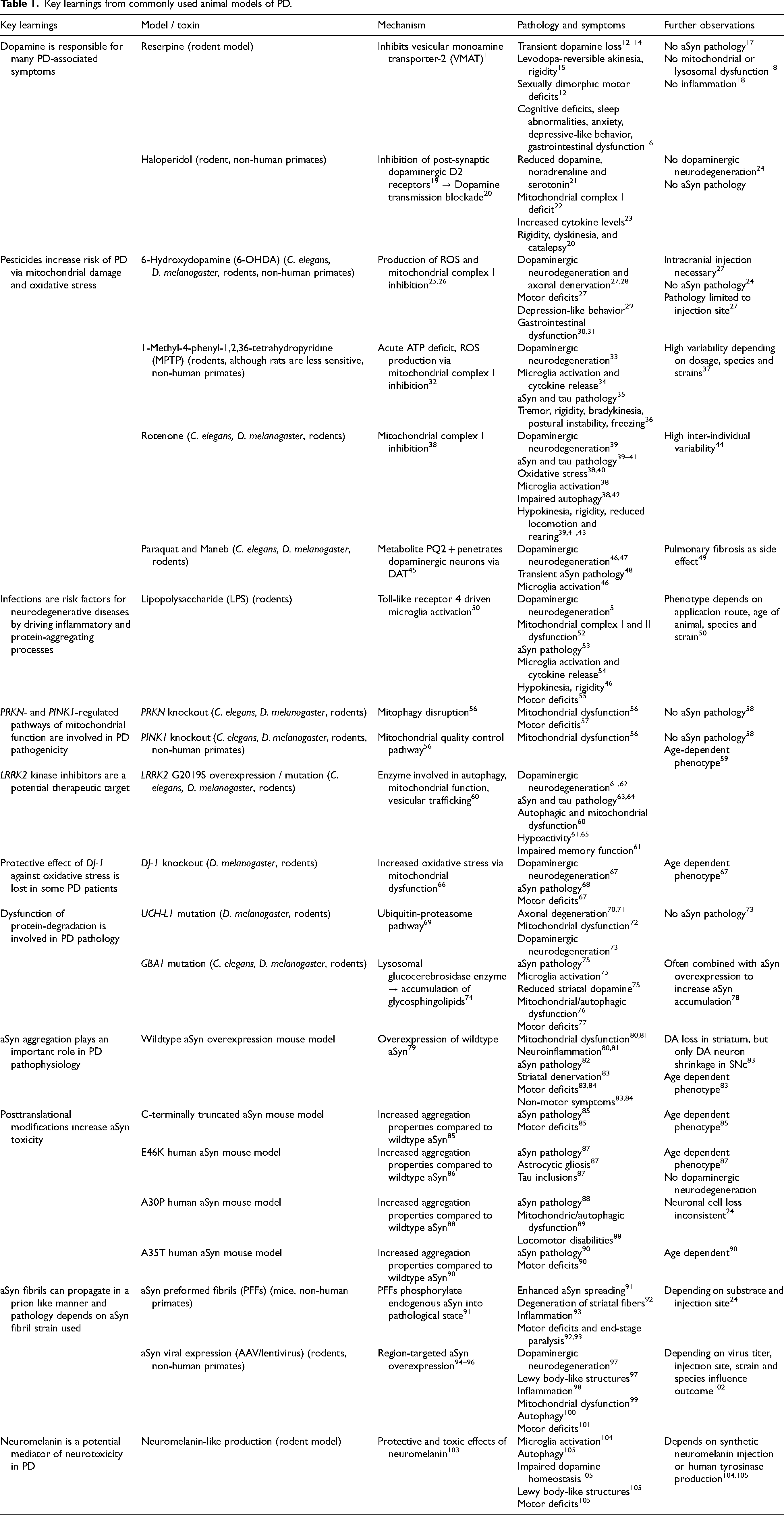
Key learnings from animal models regarding the pathomechanisms of PD
Toxins, pesticides and infections contribute to PD risk
Apart from aging, there are several risk factors that contribute to PD and its increasing prevalence. 106 According to the multiple hit theory, PD is the consequence of additive risks acquired during lifetime.107,108 These insults include genetic risk factors, but also intrauterine and peri-/postnatal infections, as well as exposure to neurotoxic herbicides or pesticides.86,109–114 While risk can be assessed in epidemiological studies, it is still not possible to accurately track pathomechanisms in patients, in absence of biomarkers, brain imaging markers, and access to brain tissue. Thus, we learned about the impact and mechanisms of risk factors from work with animal models.
A striking example comes from a major breakthrough in PD research, with the discovery that 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridin (MPTP), an impurity in desmethylprodine solutions synthesized and injected by drug addicts, induces a persistent parkinsonian syndrome in humans.113,115 As a lipophilic compound, MPTP crosses the blood brain barrier and is converted by astrocytic MAO-B into MPP+, which is selectively taken up into dopaminergic neurons via the dopamine transporter. Within mitochondria, MPP + inhibits complex I, mimicking key aspects of PD pathology.116–118
Most of these mechanisms were uncovered in mice and non-human primates, which are sensitive to MPTP toxicity, whereas rats are relatively resistant. 119 These observations led to research into mitochondrial deficiency in PD patients, and the role of oxidative stress in the pathogenesis of PD, which both are now accepted as key pathogenic pathways. Follow up work in mice and rats using pesticides such as paraquat and rotenone uncovered that the increased risk of developing PD if exposed to these environmental toxins is due to complex pathomechanisms including mitochondrial inhibition and increased oxidative stress. Of note, the risk for neurodegeneration increases if the pesticide exposure is combined with certain genetic predispositions, such as changes in the expression of the dopamine transporter.120–123 This led to re-evaluation of the use of pesticides across the world. An important aspect of the specific susceptibility of dopaminergic neurons to oxidative stress is the presence of neuromelanin, a by-product of toxic dopamine metabolism which accumulates with aging. Over-expression of tyrosinase in the rat substantia nigra has been used to induce neuromelanin deposition, 105 confirming its toxicity for dopaminergic neurons.
More recently, there has been renewed focus on the role of infections as risk factors for PD. Similar to low-dose toxin exposure, the risk increase is likely compounded over the lifetime, and very difficult to track in humans. We recently contributed to a review of the current literature, demonstrating significant evidence for viral and bacterial infections to increase PD risk. 124 Again, part of this evidence, and specifically insights into the underlying mechanisms, were derived from animal models. For instance, it was hypothesized that infections with influenza virus, West-Nile virus, picornavirus, coxsackievirus or SARS-CoV-2 could be associated with or even cause aSyn pathology.124–126 While SARS-CoV-2 is generally not a neurotropic virus, we and others demonstrated that intranasal infection in hamsters can cause persistent inflammation across the brain, accompanied by tau and aSyn pathology and loss of GABAergic interneurons, with sex-specific pathology patterns.127,128 These findings could, at least in part, explain how viruses could cause neurological symptoms in the absence of severe neuroinvasion. Repeated exposure to viruses could, similarly to environmental toxins, increase the risk to initiate neurodegenerative processes. If these processes are self-propagating, they would become unmasked as progressive neurodegenerative disease with aging.
Although the precise underlying mechanisms require further research, nearly all studies on infection and PD demonstrate a role of complex immune responses, requiring communication between the peripheral and the brain-resident immune cells across the blood brain barrier. For instance, during gastrointestinal dysbiosis, the bacterial endotoxin lipopolysaccharide (LPS) can enter the blood stream, leading to an acute peripheral immune response followed by microgliosis in the brain, which in turn may impact neuronal function.129–133 These complex mechanisms highly relevant to the human disease require an intact organism to study, and while there are significant differences in the cellular immune systems makeup, mice are established and well-characterized models to study the immune response. Especially the possibility to genetically modify specific immune cells and receptors in mice enables proof of concept studies. 134 Furthermore, testing any type of drug interfering with the immune response requires careful efficacy and safety studies in vivo in order to exclude any unexpected deleterious target or off target effects on the brain or other organs.
How aSyn may be detrimental to neurons in PD
Cytoplasmic deposits and dystrophic neurites, so-called Lewy bodies and Lewy neurites, are immunopositive for the protein aSyn and represent the histopathological hallmark of PD. 135 Lewy body pathology has been suggested to spread in the brain, and also outside the brain, during disease progression, and assessing its distribution is a common method to evaluate the stage of disease in postmortem brain tissue (Braak staging system).136,137
aSyn is a small 14 kDa and 140 amino acid long protein, and its accumulation characterizes PD as a synucleinopathy. 138 In its native form, aSyn lacks defined tertiary structure and, therefore, has been described as an intrinsically disordered protein (IDP).139,140 However, in synucleinopathies the protein forms aggregates rich in β-sheet structures that can propagate from cell to cell in a prion-like manner.141,142
Several different posttranslational modifications (PTMs) can be found in aSyn in postmortem PD brains (e.g., phosphorylation at Serine 129 (pS129), ubiquitination, C-terminal truncation, or glycation).143–146 While these observations from patient brains support an important role of aSyn in PD pathophysiology, the underlying mechanisms have been discovered and studied in disease models. In mice, it was shown that increased aSyn expression is sufficient to cause its aggregation in neurons, with concomitant neuronal dysfunction, similar to what is thought to happen in PD.82,147,148 Furthermore, overexpressing mutated familial forms of aSyn causes PD-like pathology and symptoms in mice, supporting a gain of toxic function, and the proteinopathy hypothesis.90,149
Studies in mouse models demonstrated that recombinant aSyn fibrils or aSyn aggregates/ Lewy bodies derived from human brains can propagate in a prion like manner from neuron to neuron or even from the gut to the brain, when injected intracerebrally or peripherally.92,150–152 Furthermore, work in mouse models contributed to the discovery that different aSyn fibril strains induce specific synucleinopathies. 153 These studies uncovered multiple potential pathogenic pathways involved, such as the induction of lysosomal and mitochondrial dysfunction. In addition, they provided in vivo evidence that oligomeric forms of aSyn may be the most toxic forms to neurons. 154
Lysosomal dysfunction and increased PD risk
As for all other proteinopathies, pathways associated with protein clearance are thought to play a major role in PD. For example, studies of the ubiquitin-proteasome system showed that some of its components are expressed less, and also that its catalytic activity is reduced in PD. 155 Furthermore, autophagy-lysosomal dysfunction is involved in PD pathophysiology, given that mutations in lysosomal genes, an accumulation of autophagic vacuoles, and reduced levels of lysosomal proteins are associated with an increased PD-risk.156–158 The discovery that Gaucher disease patients or carriers of mutations in the gene encoding glucocerebrosidase (GBA) have a higher risk of developing PD substantiated lysosomal dysfunction as relevant pathway. Follow up work in cell culture proposed a positive feedback loop between aSyn accumulation and lysosomal dysfunction.159–161 Lysosomal dysfunction is a common observation in toxin-induced and genetic models of PD, and has been extensively studied. Furthermore, cross-breeding of animal models of lysosomal storage disorders, with PD mouse models, demonstrated increased PD pathology, as a proof of concept. These animal models were used to develop and test novel therapeutic strategies, of which some are currently tested in clinical trials.162–167
Key learnings from animal models regarding current PD therapies
Refinement of symptomatic dopamine replacement therapy
The relationship between motor symptoms and the dopaminergic system was first demonstrated in 1957 via reserpine administration, which induced an akinetic state associated with catecholamine depletion in the central nervous system. This state could be reversed by L-DOPA administration,15,168 laying the foundation for all subsequent insights into PD pathophysiology. However, reserpine does not kill dopaminergic neurons, limiting its use to study therapeutic interventions.
The neurotoxin 6-hydroxydopamine (6-OHDA) selectively targets catecholaminergic neurons via monoamine transporters, inducing neurodegeneration.169,170 The discovery of the effects of this neurotoxin in animals was instrumental for the field. As a hydrophilic molecule, 6-OHDA cannot cross the blood brain barrier and must be injected stereotaxically—either intraventricularly, intrathecally, or intracerebrally. Intracerebral injection allows targeted, unilateral or bilateral application.171,172 Its toxic effects are thought to result from oxidative stress due to its metabolic similarity to dopamine.173,174 The classic unilateral 6-OHDA injection model produces rotational behavior after dopaminergic stimulation (e.g., apomorphine, amphetamine).175,176 As the model lacks progressive degeneration as well as aSyn inclusions, it is not suitable for developing and assessing disease-modifying therapies. However, it has been indispensable to refine dopamine replacement therapy, especially as rats and mice develop dyskinesias under L-DOPA treatment, similar to PD patients.177–179
Challenges in developing disease-modifying therapies
In contrast to symptomatic treatment, disease-modifying therapies are meant to target neurodegenerative processes, in order to halt or prevent progression.180,181 However, promising therapeutic candidates failed to reach the primary endpoints defined in the trials - improvement of motor function. 182 Strikingly, all of these compounds did, at some stage of development, show efficacy in animal models and, therefore, this lack of predictive validity is a common argument against the use of animal models. Nevertheless, the role of these models is to enable the testing of research hypothesis, and to provide the proof of concept that the drug reaches and engages its target.
In pre-clinical work, animal models that replicate the targeted signaling pathway and pathophysiology are chosen, and if the drug engages its target, these models will provide the respective positive response. As an example, models based on the injection of aSyn pre-formed fibrils (PFFs) as described above, model the formation and prion-like spreading of aSyn pathology across neurons. Drugs that directly interfere with oligomerization or misfolding of aSyn could be tested in these models. However, whether this will be sufficient to reduce the already ongoing multifaceted pathology in dopaminergic neurons of clinically diagnosed PD patients, is questionable. Likely, if the same compound is tested in a model of mitochondrial deficiency, or microglia activation, it will not result in positive outcome.
The important take home message from failed clinical trials is, that neuroprotection in PD will require treating very early in disease progression, long prior to clinical diagnosis, and/or developing strategies of drugs that target several pathogenic pathways, or combining drugs with complementary, additive or even super-additive effects. In any case, the animal models used need to be carefully chosen to replicate the targets to be engaged. 84 A single model should not be expected to encompass such a multi-system, multi-faceted and multi-mechanistic disease, it should at least replicate nigrostriatal dysfunction and motor symptoms to predict main clinical endpoints, together with aSyn pathology and microgliosis.82,183,184
In recent years, molecular changes were proposed to be the more valid endpoint compared to behavioral signs, however, this did not lead to success in clinical trials. Instead, it may be most important to understand which molecular pathway alteration in which brain region is linked to a specific behavioral readout. In toxin induced models, this relationship is straight forward, e.g., dopamine loss is linked to motor deficits in the 6-OHDA model described above. In more slowly progressing synucleinopathy models with pathology across peripheral and central neurons, involving multiple pathogenic mechanisms, this direct link between the construct and face validity is more challenging to decipher.82,183,184
Another often overlooked aspect of the role of animal models are the many drugs that do not make it to the clinics because their effects were deleterious in animal models. If the prediction of clinical benefit for a drug target is built on an incomplete picture of its role in neuronal function, the drug could have unexpected deleterious on-target effects. Consistently, we recently showed that mice over-expressing aSyn replicated the clinical failure of venglustat, thereby providing possible mechanisms for the observed worsening of motor symptoms in patients. 166 The bulk of experimental evidence for venglustat to move to the clinics was derived from advanced in vitro models, and the results were valid in these young cells accumulating lysosomal substrates. However, they did not translate into clinical benefits.
The reasons why the various strategies tested thus far showed no improvement of motor function in PD patients can be manifold and are, for the most part, still unclear. Nevertheless, failed trials also offer tremendous opportunities for learning. For instance, they suggest that the impact of tested therapeutic targets on the degeneration of dopaminergic neurons at the stage of disease when the clinical trial is conducted may have been overestimated. The degenerative processes may have been too far advanced: e.g., mitochondrial dysfunction, lysosomal dysfunction, build-up of aggregated proteins, inflammatory processes. In addition, failed trials suggest that halting one signaling pathway in one of these processes is likely not sufficient to improve dopamine neuron function such that the clinical outcome improves significantly.
Studies involving animal models remain instrumental for moving into clinical trials, even if the FDA is making some exceptions for some conditions. Currently there are a number of Phase 2 and 3 trials with PD therapeutics in the planning stages or ongoing, 185 with a few disease-modifying strategies. The diagnosis of PD has recently shifted the focus on underlying disease biology. 186 If quantifiable biomarkers and brain imaging will become applicable as clinical endpoints, we will be able to further refine in vivo efficacy studies to increase predictive validity and to reduce the numbers of required animals.
Ex vivo and in vitro strategies for assisting and reducing the use of animals
Strategies for reducing or replacing animal experiments are evolving in the field of PD research. While cellular models cannot reproduce the broad complexity of PD pathophysiology, they enable modelling the cellular phase of the disease via specific gene manipulations, and the study of various molecular processes such as aSyn pathology, oxidative stress, autophagy, or apoptosis. Importantly, they are also suitable for high throughput screenings.186,187 Therefore, they offer a highly valuable toolbox for assisting and guiding animal experiments, thereby increasing our understanding of disease mechanisms and ultimately enabling the identification of suitable targets for therapeutical intervention (Figure 1). By rapidly screening therapeutic targets and candidate molecules instead of using animals for these efforts, in vitro methods ease ethical concerns raised by the use of experiments in mammalian systems, while also contributing to reduce cost and to save time.
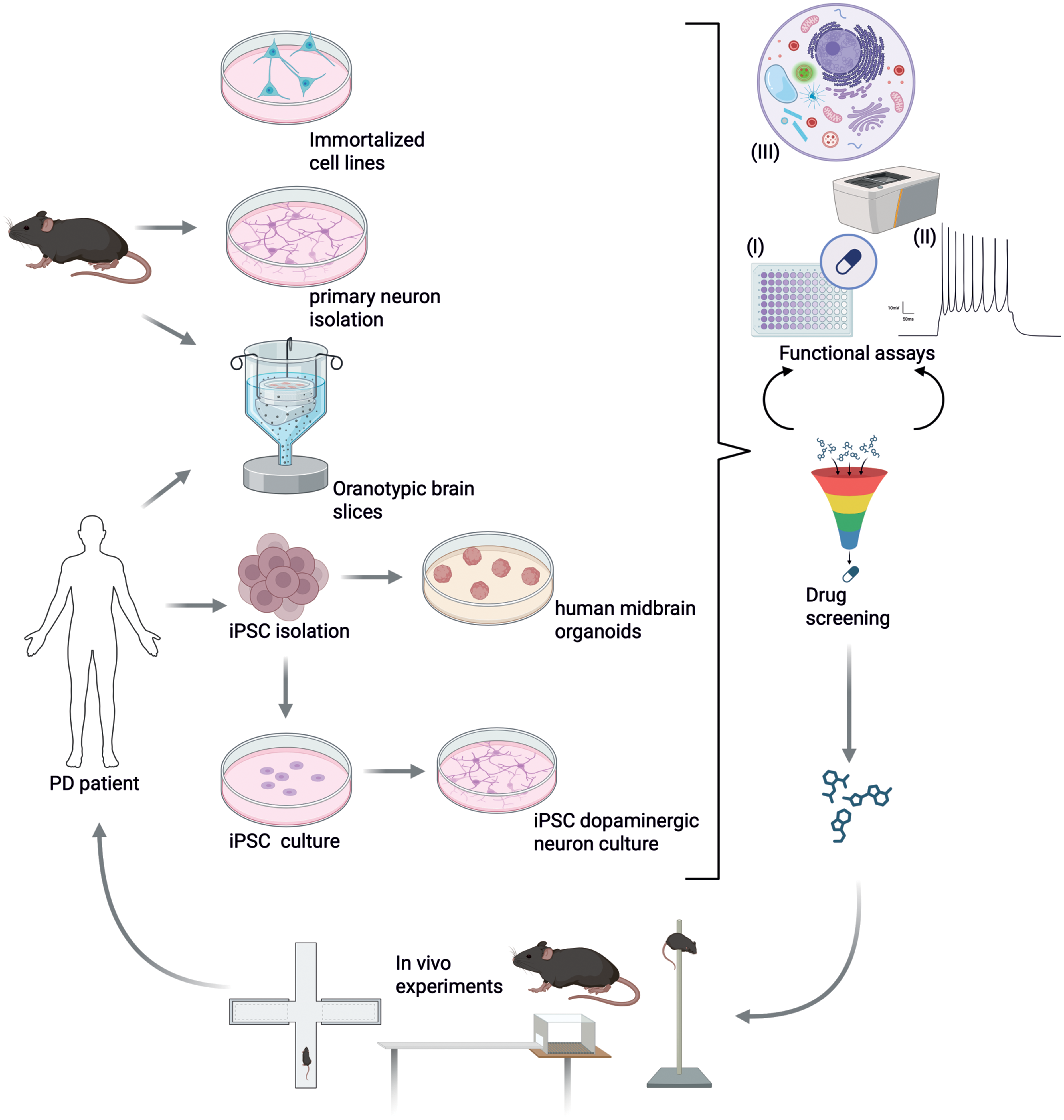
Example of combined uses of in vitro and in vivo models during drug screening. In vitro and ex vivo PD models include immortalized cell lines, primary neurons, 3D-structured organotypic brain slices, iPSCs and organoids. Especially, regarding cellular and molecular mechanisms of disease progression, focusing solely on cells instead of whole organisms enables a deep analysis of cellular pathways. Functional assays to measure (I) cell death in context of neurotoxicity due to protein accumulation or dopamine-derivates, (II) neuronal firing rate to assess functionality of neurons, and (III) tracking of organelles to study protein degradation or mitochondrial dysfunction more precisely. Evolving in vitro methods are suitable for high throughput screening of drugs since access to cell models is easier and ethical concerns are avoided. They should be seen as additional tools to investigate mechanisms of PD and identify suitable targets for therapeutic intervention. Animal models will always be a crucial part in PD research, to determine progression of motor symptoms and long-term effect of treatment options using motor tests. Figure created with BioRender.
In vitro models can be very useful especially when studying cellular mechanisms and cellular pathways related to cytotoxicity or neuronal network degeneration during PD progression. 188 Moreover, novel in vitro assays also allow quantification of neuronal firing rates, neuronal axon and network growth, lysosomal tracking, and mitochondrial functionality. All of these approaches enable in-depth investigations of cellular pathways, easily overlooked when using a whole organism. For example, immortalized cell lines that resemble human dopaminergic neurons and primary neurons are useful tools to study cell toxicity and perform high throughput screening of drugs. However, these models have several limitations including the cancerous properties of immortalized cell lines and the lack of authentic dopaminergic characteristics.187,189 And due to the two-dimensional and single-cell culturing method, important cell-cell-interaction is lost. Therapeutic intervention or prevention of disease progression to this day still resembles the main challenge in PD and focusing on one subtype of cells is likely insufficient to predict clinical efficacy. 190
Organotypic brain slices
Tissue from genetically modified animals or PD patients can be cultured in an in vitro system called organotypic brain slices. This method preserves the physiological cell-to-cell interaction, by maintaining the architecture. Moreover, the simultaneously culturing of functionally related brain areas offers a unique tool to study aSyn spreading and seeding abilities.191,192 For example, when injecting PFFs into the dentate gyrus of organotypic hippocampal brain slices, the aggregation of aSyn and its spreading to CA3 and CA1 regions can be observed.193,194 To study dopaminergic circuitries, organotypic brain slices of the substantia nigra and striatum are established. 195 Moreover, the important interplay of neurons and glial cells during PD progression can be observed as well as neuronal activity measured via multielectrode array compatible culture methods.196,197 Working with neurotoxins like MPTP and 6-OHDA, frequently used in animal models and in cellular models, has the disadvantage that aSyn pathology is missing and dopaminergic neurodegeneration is achieved only via oxidative stress induced by the toxin. 198 Mechanical transection of nigrostriatal pathways which initiates dopaminergic neurodegeneration is another method to induce neuronal loss in organotypic brain slices. 199 However, also this method lacks aSyn pathology.
Organotypic brain slices are often isolated from early postnatal mice or rats, but nowadays also protocols for adult mice/rat brain exist.200,201 This allows preclinical testing in aged material with observable and measurable neuronal network activity over an extended period. Surgically resected human tissue and even postmortem tissue from humans can be used as donors.202–204 This represents a powerful method to observe disease-associated pathology during culture. 203 Overall organotypic brain slices obtained from animal models are useful additions to the in vivo studies and offer opportunities for human-tissue based preclinical drug development. 203
IPSCs
Patient-derived inducible pluripotent stem cells (iPSCs) can be generated from cells taken, for example, as a skin biopsy of PD patients. These cells can be reprogrammed to embryonic stem cells with the complete genomic background of a patient and then used as cellular model with patient specific cells. 205 Several protocols exist to differentiate stem cells into dopaminergic neurons, making it a suitable method for PD research.206,207 IPSCs represent a very powerful tool, as it holds the ability of high throughput screening individualized to a patient. 205 This in vitro strategy is often used to investigate how PD-associated mutations affect cellular pathways, since PD patients with known mutations were mostly used as donors.208,209 Recently, sporadic PD patients are used as donors to understand the vulnerability of their neurons to the disease seeking to find common altered pathways which can be targeted by a drug. 210 For example, iPSCs from PD patients show abnormal accumulation of aSyn and downregulation of antioxidative pathways, making them sensitive for oxidative stress. 210 Although sporadic PD patients do not show a mutation associated with increased PD risk, iPSCs generated from this subgroup of patients show epigenetic changes which could add to their increased susceptibility. 209 Increased oxidized dopamine levels and oxidative stress are also found in iPSCs generated from patients with mutations in genes associated with PD risk like SNCA, LRRK2, PINK1/Park2, and VPS35.208,211
Overall, iPSCs do not only offer individualized screening for suitable therapeutical options, but they may also be suitable treatment options in the future. 212 Allele-specific genome-targeted knock-in and knock-out via the CRISPR-Cas9 system allows regulation of endogenous gene expression and might lead to disease-resistant iPSCs as implants to replace the lost dopaminergic neurons.213–216 A pitfall of iPSCs cultures is that they are for the most part lacking non-neuronal cells, such as microglia, astroglia or endothelial cells. There is ongoing research to overcome this limitation by co-culturing or creating complex organoids. Another criticism raised specifically for studying neurodegenerating diseases is that these cells do not replicate the impact of aging, as they remain relatively young neurons. However, in combination with other models iPSCs will further develop into a key in vitro system to develop therapy especially if specific genetic subtypes carrying certain mutations or risk alleles are targeted.
Organoids
The absence of 3D structure in in vitro systems means that the typical cell-cell-interactions of the brain, and the formation of a physiological cytoarchitecture, cannot be maintained. This is aggravated by the fact that the central nervous system is composed of several cell types that are usually not present in 2D culture systems. 3D systems based on iPSCs are being frequently used as so-called organoids, that self-organize to mimic the 3D structure of the brain. 217 This system allows the highest neuronal differentiation ex vivo and resembles a neuronal network with different subtypes of cells present in the brain.218–220 Midbrain-like organoids are described to contain neuromelanin-positive, dopaminergic neurons and to form a functional, mature neuronal network. 221 Using iPSCs from human PD patients allows the differentiation of brain-like structure that mimic the key hallmarks of PD, like decreased dopaminergic neurons. 222 PD-specific aSyn pathology can be recapitulated when cells are derived from patients with SNCA triplications. Elevated levels of aSyn and phosphorylation of the protein was detected, while the organoid expressed increase senescent markers and reduced dopaminergic neurons.223–225 As organoids also recapitulate rhythmic electrical activity, they are suitable to quantify alterations of neuronal activity, not only neuronal cell loss. 226
Thus far organoids are focused on specific cell types, but efforts are made to create multi-cell type models that contain neurons, glia and aspects of the blood brain barrier. For example, organoids can be co-cultured with microglia precursor cells which also induces the development of vascular structures. 227 Microfluidic chambers allow active perfusion of the necrotic organoid core and enable observation of cell-vessel interaction.228,229 However, comparison of cortical cells and cortical organoids via high-throughput single-cell RNA sequencing revealed reduced subtype resolution due to lack of maturation. 230 Similar to iPSCs, organoids model the embryonic development of the brain and lack age-dependent characteristics as well as myelination and microglia and oligodendroglial migration.231–233 Not only aging but also maturation of synapses, dendrites and axonal projections are missing, making studies on nigrostriatal circuitries impossible.234,235 Extending the culture period up to 250 days allows maturation of the organoid, but immense costs and low reproducibility restrict the utilization of this method. 232 Pharmacologically induction of aging in organoids alters cellular pathways which could be misleading when studying age dependent diseases like PD.236,237
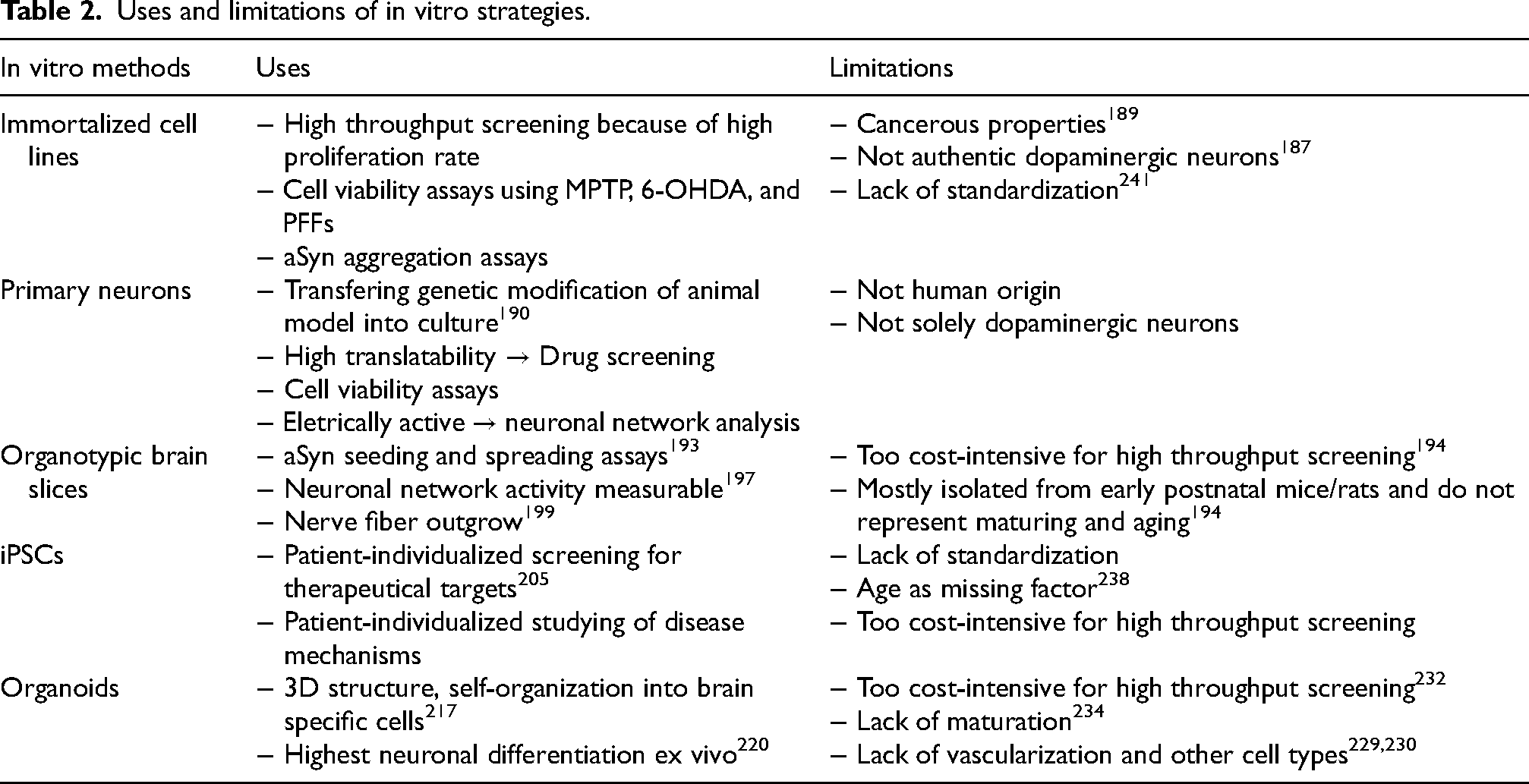
Uses and limitations of in vitro strategies
In vitro strategies are rapidly developing field in neuroscience and their continuous improvements will increase their usability in PD research. Key examples for uses and limitations are summarized in Table 2. Out of all limitations, the poor translational value remains one of the biggest obstacles of in vitro studies at the moment. Although in vitro models are instrumental for mechanistic/molecular studies, it is important to consider that, for example, immortalized cell lines carry cancerous characteristics that must be considered. 189 Even iPSCs, which from a genetic point of view, closely resemble the human they were generated from, lack the highly-relevant factor of age. Due to the reprogramming, cells are set back to the chronological age of almost foetal, which is observable in an increase length of telomerases. 238 Effort is put into establishing iPSCs that express aging marker, but they still exhibit different mitochondrial arrangements and lower oxidative stress compared to adult cells.239,240 Not only immortalized cell lines but also primary neurons and iPSCs show high variability among clones which makes comparison between studies and experiments hard. The described variabilities appear unrelated to PD-phenotype or genotype and is most likely due to differences in maintenance and isolation protocols. 241 Bridging knowledge gaps by combining in vitro and in vivo experiments might bring up promising therapeutical targets. 242 Beyond that, in vitro methods can be used strategically to reduce costs and ease ethical concerns when screening for drugs.
Uses and limitations of in vitro strategies.
As modelling of PD is based on genetic or toxic manipulation in cell culture systems and in animal models, the in vitro methods suffer from similar limitations as the accompanying in vivo experiments. This includes the possibility to measure downstream alterations of the modification not associated with PD pathology or to induce compensatory mechanisms in the cells, which do not resemble the disease. When working with immortalized cell lines, the immortalization changes the cell genotype and translation to the human situation might not be reliable. 190
Lastly, one major drawback of in vitro and ex vivo strategies is the lack of vascularization, for example in organoid models. Crosstalk happening at the blood-brain-barrier and immune invasion are described to play a key role in PD progression.187,243 When blood flow is missing, detecting possible biomarkers for blood analysis can only be achieved by measuring substances, secreted into the cell culture media. 244 While organotypic brain slices show a network of laminin-positive brain capillaries, they are not functional in culture. 245 Nonetheless, organotypic brain slice culture represents the closest to in vivo to study secretion of vascular molecules and response to angiogenic substances in culture.246,247
Minding the limitations and uses, in vitro models should be included in experiments in PD research to improve our understanding of the disease and improve replacement methods. Overall, PD might be a cellular disease, indicated by functional loss of several cellular pathways including autophagic dysfunction, mitochondrial dysfunction, ER alterations. 248 To reduce cost issues and low translation, more research into innovative strategies is necessary. 187
Conclusion: Do we still need animal models in PD research?
One of the major drawbacks of in vitro models is that they lack the ability to reproduce the complex architecture of the brain, its functional readout, and its connection to other organs. And while it is well-described and recognized that each in vivo model also lacks major parts of disease characteristics, they do not have these limitations. However, the advantages of emerging in vitro methods discussed above show the potential of reducing animal experiments in PD while simultaneously opening new possibilities to screen for therapeutics. Knowledge gaps in PD research might not be filled by sticking solely to animal models, but without them we will not be able to develop safe and efficacious drugs. Currently, aSyn pathology is the most investigated and tested drug target for disease-modification in PD, and while specific mechanisms can be further studied and validated in in vitro models, the complexity of its spreading and down-stream pathology requires a complete brain with all cell types connected in a functional neuronal network for finite conclusions and drug efficacy testing. New methods that simplify the complex disease at the level of single cells, might offer opportunities to detect cellular mechanisms not yet discovered. 190 Nonetheless, animal experiments will be needed in PD research to detect behavioural deficits replicating main clinical endpoints, which cannot be mimicked by in vitro methods. 188 Ultimately, we argue that multimodal approaches, combining the findings of different in vitro and in vivo models, will more likely lead to the identification and validation of the long-sought disease modifying strategies for treating PD and related disorders.
Footnotes
Acknowledgements
The authors are grateful to the many colleagues which, in past, present and future, work tirelessly to improve treatment of Parkinson's disease, using in vitro and in vivo models, thereby certainly adhering to 3R principles, which is part of good laboratory practice and ethical guidelines in research.
Ethical considerations
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. No approval was required from the local ethical committee for this review article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by intramural funding from the University of Veterinary Medicine Hannover.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Datasets/data availability statement
The raw data of this study are available on reasonable request from the corresponding author.