
Abstract
Background
Huntington's disease (HD) is a neurodegenerative disorder caused by CAG expansions in the Huntingtin (HTT) gene. Due to its non-specific and variable phenotype, diagnosis requires clinical assessments and genetic testing. In the Caribbean, the genetic etiology of HD is underexplored due to the unavailability of genetic testing.
Objective
We investigated whether 32 participants from four multigenerational families from Trinidad and Tobago (T&T) presenting with Huntington-like symptoms carried HTT CAG expansions, and whether CAG length was related to decreasing age of onset with each generation.
Methods
Participants were genotyped using triplet repeat primed PCR followed by previously-established fragment analysis and a nanopore sequencing based method with a custom bioinformatics workflow.
Results
All symptomatic participants carried HTT CAG expansions (42–57 CAGs), confirming HD. Among participants aged 20–65 years (n = 24), clinical and genetic diagnoses were concordant for 22 participants (13 symptomatic with 42–57 CAGs, and nine asymptomatic with 13–27 CAGs). Two asymptomatic participants aged 22 and 43 years carried 46–47 and 37–39 CAGs, respectively. Among eight participants <18 years, one symptomatic 16-year-old carried 49–50 CAGs, and seven are currently asymptomatic (three with 50–52 CAGs, and four with 14–17 CAGs). In three families, decreasing age of onset and increasing CAG length were observed in each successive generation. Methods were highly correlated (R2 = 0.998).
Conclusions
We demonstrated the application of nanopore sequencing with a custom bioinformatics workflow to estimate the size of HTT CAG repeats. This is the first genetic report of HD in T&T, among limited records in the Caribbean.
Keywords
Introduction
Huntington's disease (HD) is an inherited, autosomal dominant, progressive neurodegenerative disorder of the central nervous system.1,2 Clinical manifestations of the disorder include motor, cognitive, and psychiatric symptoms. 1 HD is associated with a CAG triplet repeat expansion in exon 1 of the Huntingtin (HTT) gene on chromosome 4p16.3. 3 HTT alleles consisting of ≤35 CAG triplet repeats are not associated with HD development. An expanded gene sequence containing ≥36 CAG triplet repeats is associated with HD. HD is fully penetrant when there are ≥40 CAG repeats and shows variable penetrance for alleles with 36–39 CAG repeats.3,4
The age of onset of HD is described as the age at which a carrier of the mutated HTT gene begins to develop unequivocal signs of the disorder. 5 The primary determinant of the age of onset of HD is the number of CAG repeats in the mutant HTT allele and there is an inverse relationship between the number of repeats and the age of onset. 6 Symptoms of adult onset of HD typically manifest between the age of 30–50 years and death occurs by 15–20 years after their appearance. 7 Larger expansions of the mutation (≥60 CAG repeats) are often associated with earlier/juvenile onset of HD and symptoms usually manifest at age 20 years or younger.8,9 While the number of CAG triplets in the abnormally expanded region of the gene can increase, decrease or remain stable when transmitted to offspring, transmission from fathers shows a strong expansion bias in each generation in a family with the confirmed mutation. 10
Trinucleotide repeat disorders, including HD, are associated with phenotypes that are usually nonspecific and variable, and so cannot be accurately diagnosed by clinical assessments alone. Molecular diagnostic tests must be employed to determine the underlying genetic aberration and estimate the number of CAG triplet repeats. The American College of Medical Genetics and Genomics indicates that triplet repeat primed PCR (TP-PCR) can be used as a primary molecular diagnostic method, though this is specifically recommended as a method for detecting long repeats that might be missed using methods that involve two repeat-flanking primers. 11 TP-PCR amplicons can be analyzed by fragment analysis or sequencing to achieve an accurate HTT genotype. 12 TP-PCR coupled with fragment analysis by capillary electrophoresis is the preferred established combination for genotyping triplet repeat alleles. 13 Accurately sizing and identifying true homozygous and heterozygous alleles are made easier by counting the number of electrophoretic peaks as they appear on an electropherogram, as opposed to visualizing DNA patterns on denatured polyacrylamide and agarose gels.14,15 Less clustered peaks are produced for normal alleles while a much larger, continuous expanse of peaks with increasing sizes and decreasing peak heights indicate expanded repeats. 13 TP-PCR also eliminates the need for labor-intensive Southern blotting to confirm very large repeat expansions.16,17 However, this method is still relatively low-throughput and relies on the use of a control sample with known genotype for accurate sizing, which is not always available.
High throughput next generation sequencing (NGS) based methods that sequence across entire TP-PCR amplicons have the potential to give exact counts of CAGs without the need for a control with known genotype. This overcomes the limitations of the established fragment analysis method. An NGS platform that allows long and flexible read lengths is ideally suited for accurately genotyping alleles that differ by size across and within individuals, such as those underlying HD and other triplet-repeat expansion diseases. 9 One such long and flexible read NGS platform is the Oxford Nanopore Technology (ONT) sequencing platform, which is emerging as a viable diagnostic tool for various triplet repeat disorders, including HD. Recently, it has been used with varying approaches in a handful of studies. One study utilized the “read until” functionality for diagnosing a list of known repeat expansion diseases without prior target genomic region enrichment. 18 Another study used nanopore sequencing to determine HTT haplotypes by performing long-range sequencing across the region. 19 A third study used nanopore sequencing to overcome PCR drop out of very long repeats which may go undetected. 20 It should be noted that highly accurate short-read, fixed read length Illumina and long-read PacBio platforms have also been used to sequence TP-PCR amplicons, but with variable success. 21 Comparatively, nanopore sequencing exhibits a relatively high base calling error rate compared to other NGS platforms particularly due to indels. 22 Still, the utility of nanopore sequencing downstream of TP-PCR as a method for accurate HTT CAG genotyping is yet to be established.
HD diagnosis necessarily involves clinical assessment along with genetic testing.7,23 Consequently, HD prevalence appears higher in developed countries, where genetic testing is more accessible, than in developing regions with limited testing resources.9,24 As a result, globally diverse populations from lesser-developed countries remain underrepresented in global HD databases. Over the past decade, increased reporting has revealed a rise in HD frequency worldwide. A 2022 review estimated the global incidence at 0.48 per 100,000 person-years and the pooled prevalence at 4.88 per 100,000 persons between 2010 and 2022. 25 Yet, the only record of HD affecting the population of Trinidad and Tobago (T&T) was documented in 1963. 26 At that time, the diagnosis was performed solely by clinical assessment of Huntington-like symptoms (motor, behavioral and psychiatric disorders) that affected members of three families. To date, no statistical data on the prevalence of HD affecting the Trinidadian population or the wider Caribbean population, have been added to scientific records. This is because of limited capacity in this region to perform genetic testing for the disorder. Nevertheless, genetic diagnosis in understudied regions is required to enable equitable access to novel gene-based therapies for HD.
The Caribbean's genetic landscape is richly diverse due to its complex colonial history but remains underexplored. The population of T&T comprises 1.3 million people of which ∼35% are Indo-Trinidadian (descendants of North Indians brought to the Caribbean under indentureship during British rule), ∼34% Afro-Trinidadian (descendants of West Africans brought to the Caribbean via the transatlantic slave trade). The remainder of the population comprises individuals of mixed ancestry, and small minorities of other ethnic groups including Chinese, Syrian/Lebanese, and Whites. 27 The genetic basis of HD in this population has not been previously characterized. As an initial investigation, we sought to determine whether canonical HTT CAG repeat expansions underlie the observed HD phenotypes in T&T. Given the established relationship between CAG repeat length and disease manifestation, TP-PCR was employed as a targeted, efficient approach to detect canonical alleles, including large expansions that may be present in our cohort. Specifically, we aimed to (i) detect canonical HTT expansions in multigenerational families presenting with HD-like symptoms, (ii) assess concordance between clinical and molecular diagnoses obtained using different genotyping methods, and (iii) examine whether intergenerational repeat expansion was associated with earlier disease onset. We also evaluated nanopore sequencing of TP-PCR amplicons with a custom bioinformatics workflow as a potential alternative to fragment analysis for estimating CAG repeat size in HD families from T&T.
Methods
Ethics statement
Ethics approval was obtained from the Campus Research Ethics Committee of The University of the West Indies (UWI), St Augustine, Trinidad on July 29, 2019 (CEC1176/06/09) and from the Eastern Regional Health Authority, Trinidad and Tobago (ERHA-REC:027/08/2019). Prior to this study, symptomatic participants were aware of their own clinical diagnoses consistent with HD, and asymptomatic participants were also aware of a clinical diagnosis consistent with HD in their family members. There is currently no standard protocol for accessing genetic testing and further, returning genetic testing results to patients in the public health system in this country. Moreover, genetic counsellors are not available in the public health system in T&T. Nevertheless, results were returned to participants via the collaborating physician, and with discussion with a human geneticist, if requested. The sizes of participants’ HTT CAG repeats were confirmed by two independent molecular methods before communicating results to the participants by the collaborating physician.
Study participants and clinical diagnosis
A total of 32 participants from four Trinidadian families (families 0040, 0041, 0042, and 0044) were included in the study after providing written informed consent. Participants from families with a clinical diagnosis of HD-like symptoms were identified by a collaborating physician and recruited either at the Sangre Grande Hospital in August 2019 (family 0040) or at a Huntington's Disease Symposium held at The University of the West Indies, St Augustine in February 2020 (families 0041, 0042 and 0044). The clinical diagnosis for HD of each participant was assessed by the collaborating physicians based on the clinical assessment parameters including motor, cognitive, behavioral, functional and independence assessments as defined by a modified Unified Huntington's Disease Rating Scale.
Sample collection and DNA extraction
Demographic and clinical data, including self-reported ancestry, were collected from participants by means of a closed-ended questionnaire. Progeny software (https://www.progenygenetics.com/) (Progeny Genetics LLC, USA) was used to construct family pedigrees based on participant responses. A blood sample was also collected from each participant. In instances where a blood sample was difficult to obtain either due to physical challenges with the participant or for children, a saliva sample was obtained instead. Data and samples were de-identified for subsequent analysis. Each participant was given a unique Patient Identification number (PID)
Blood samples were collected in 4 ml BD Vacutainer K2EDTA tubes (Becton Dickinson, USA) and saliva samples were collected in OG-600 Oragene DNA Saliva Self-Collection Kits” (DNA Genotek, Canada) following the manufacturer's instructions. Genomic DNA was extracted from blood using the QIAamp® DNA Blood Mini Kit (Qiagen, USA) and ReliaPrepTM Blood gDNA Miniprep System (Promega, USA) according to the manufacturer's protocols. Genomic DNA was extracted from saliva samples using the prepIT L2P Manual Purification kit (DNA Genotek Inc., Ottawa, Canada) according to the manufacturer's protocol. DNA quantity and quality were determined by standard methods using the NanoDrop™ 2000 Spectrophotometer (Thermo Fisher Scientific, USA) and 1% agarose gel electrophoresis.
TP-PCR primers
All PCRs used in this study were TP-PCR using the previously published primers listed below.
13
Forward Primer: 5’-ATGAAGGCCTTCGAGTCCCTCAAGTCC-3’ Labelled Forward Primer: 5’-/6-FAM/ATGAAGGCCTTCGAGTCCCTCAAGTCC-3’, with its 5’-end labelled with carboxyfluorescein (6-FAM). Reverse Primer: 5’-CGGTGGCGGCTGTTG
TP-PCR and Sanger sequencing
TP-PCRs of total volume 25 µl were prepared with 0.5 µl of 10 µM unlabeled forward and reverse primers listed above (IDT, USA), 12.5 µl GoTaq® Green Master Mix (Promega, USA), 2 µl of 1–3 ng/µl of extracted DNA and 9.5 µl nuclease-free water (Promega, USA). Amplification was performed on the MultiGene OptiMax thermal cycler (Labnet International, Taiwan) under the following conditions: initial denaturation at 95° C for 5 min, followed by 35 cycles of denaturation at 94° C for 1 min, annealing at 64° C for 1 min, extension at 72° C for 2 min, and a final extension at 72° C for 15 min.
PCR products were separated based on their molecular weight by gel electrophoresis at 80 Volts for 1 h and 45 min, on a 1.5% (w/v) agarose gel. Each band was cut using a sterile, stainless-steel blade, purified using the Wizard® SV Gel and PCR Clean-Up System (Promega, USA) following the manufacturer's instructions and sent for Sanger sequencing at Macrogen Inc. (Seoul, South Korea) using their standard bi-directional sequencing service.
TP-PCR and fragment analysis
TP-PCRs of total volume 25 µl were prepared with the following reagents contained in the “GoTaq G2 Hot Start Polymerase” kit (Promega, USA): 5 µl of 5X colourless buffer, 1.9 µl of 25 mM MgCl2, 0.25 µl of 5 U/µl GoTaq G2 Hot Start Polymerase, and 0.8 µl of dNTP Mix with each nucleotide at 10 mM (Promega, USA), 0.5 µl of 10 µM labelled forward and reverse primers as listed above, 2 µl of 1–3 ng/µl of extracted DNA and 13.05 µl nuclease-free water (Promega, USA). PCR cycling conditions were as above.
Fragment assay and data analysis using the GeneMapper software were performed at the Genomics Core Facility at the University of Utah (Salt Lake City, Utah). One µl of each PCR product was mixed with 1 µl GeneScan 500 LIZ internal size standard (Thermo Fisher Scientific, USA) and 9 µl of HiDi formamide (Applied Biosystems). The mixture was heated for 2 min at 95° C and placed on a cold block for 2 min. Fragments were resolved by capillary electrophoresis on an ABI 3730 DNA Analyzer (Applied Biosystems) followed by data analysis. For the purpose of assay interpretation, the most prominent peak/s generated by the GeneMapper software were used to determine the number of CAG repeats and the continuous stuttered peaks were used to determine expanded CAG alleles in heterozygous samples. The non-CAG bases of the primer sequences (42 bases) were subtracted from each PCR amplicon length (boxed numbers seen in the electrophoregrams in Supplemental Figure 3) and the remaining value was divided by 3 to calculate the number of CAG repeats for each sample. We were unable to obtain a control with a known number of CAG repeats, and therefore were unable to convert the length of each PCR fragment confidently to an absolute number of CAG repeats. However, one known true homozygous normal sample was distinguished from the expanded alleles by the absence of a stuttered pattern that denoted an expanded allele.
TP-PCR, library preparation, nanopore sequencing, and analysis
TP-PCR mixtures and amplification were performed as described above in “PCR amplification for fragment assay and data analysis” but using un labelled forward and reverse primers instead.
Amplicon library preparation was conducted in the Molecular Genetics and Virology Lab in the Department of Preclinical Sciences at the Faculty of Medical Sciences, UWI (St Augustine, Trinidad and Tobago), following the manufacturer's instructions provided for the “Ligation sequencing amplicons (SQK-LSK109)” and “Native Barcoding Expansion 1–12 (EXP-NBD104) and 13–24 (EXP-NBD114)” protocol (Oxford Nanopore Technologies, UK). Modifications to the protocol include: the use of 2.4 µl Ultra II End Prep Reaction Buffer and 1 µl Ultra II End Prep Enzyme Mix for 30 µl of each PCR amplicon (end preparation); 1.25 µl NBXX barcode and 8 µl Blunt/TA Ligase Master Mix for a reduced volume total (native barcode ligation). 7.35 ng of the library was loaded onto an R9 flow cell and run on a GridION X5 Mk1 sequencer (Oxford Nanopore Technologies, UK) for 72 h.
Raw fast5 files retrieved from the GridION were converted to FASTQ files using Dorado Basecall Server 7.2.12 (Oxford Nanopore Technologies, UK). Each sample produced multiple FASTQ files which were concatenated into a single FASTQ per barcode and the quality was checked using PycoQC v2.5.2 (https://github.com/a-slide/pycoQC). General matrices of the raw reads were obtained using Nanoplot v1.41.0 (https://github.com/wdecoster/NanoPlot). Reads were filtered only to include those greater than 20 bp and with a quality score of 9 using Nanofilt v2.8.0 (https://github.com/wdecoster/nanofilt). Metrics of the filtered reads were checked using Nanoplot v1.41.0.
A custom designed Python script was implemented to perform the following operations (available at https://github.com/haraksingh-lab-uwi/2025-molecular-diagnosis-hd-trinidad or DOI 10.5281/zenodo.15587961). The .fastq files for each sample containing high-quality forward and reverse reads were inputted. The Smith-Waterman alignment algorithm was run with a command to allow zero mismatched bases (SW0) and then allow up to ten mismatched bases (SW10) for each primer to identify reads with the following sequence format “5’- forward primer- any sequence- reverse primer complement −3’” and “5’- reverse primer- any sequence- forward primer complement −3’.” An output file containing a table with read lengths and frequencies of the sequences identified above was generated. These data were plotted on histograms of frequency versus read length. The most frequent read lengths were used as a proxy for the size of the alleles. Up to 10 mismatches were allowed to include sufficient reads for analysis because of the known high base-calling error rate of ONT. Linear regression and Bland-Altman analyses were performed to assess the agreement between the number of CAG repeats obtained from the nanopore sequencing and fragment analysis methods.
Classification of genotypes for HD
CAG sizes were estimated based on calculations from fragment lengths or read lengths. Participant genotypes were classed into categories of normal, intermediate, reduced-penetrance and full-penetrance for HD by binning the number of CAG repeats obtained from two molecular diagnostic methods based on previously established categories as follows: ≤26 CAG repeats classified as normal genotype and will not develop HD, 27–35 CAG repeats classified as intermediate HD genotype and will not develop HD, 36–39 CAG repeats classified reduced-penetrance HD genotype and may or may not develop HD, and ≥40 CAG repeats classified as full-penetrance HD genotype and will develop HD. 11
Results
Participant demographics and clinical diagnosis
A total of 32 participants (ages 7–65 years) from four families were included. Of these, 14 (ages 16–65) were clinically symptomatic and the remaining 18 (ages 7–53) were clinically asymptomatic. There were eight participants <18 years. All were clinically asymptomatic except for one symptomatic 16-year-old (Figure 1A-D).
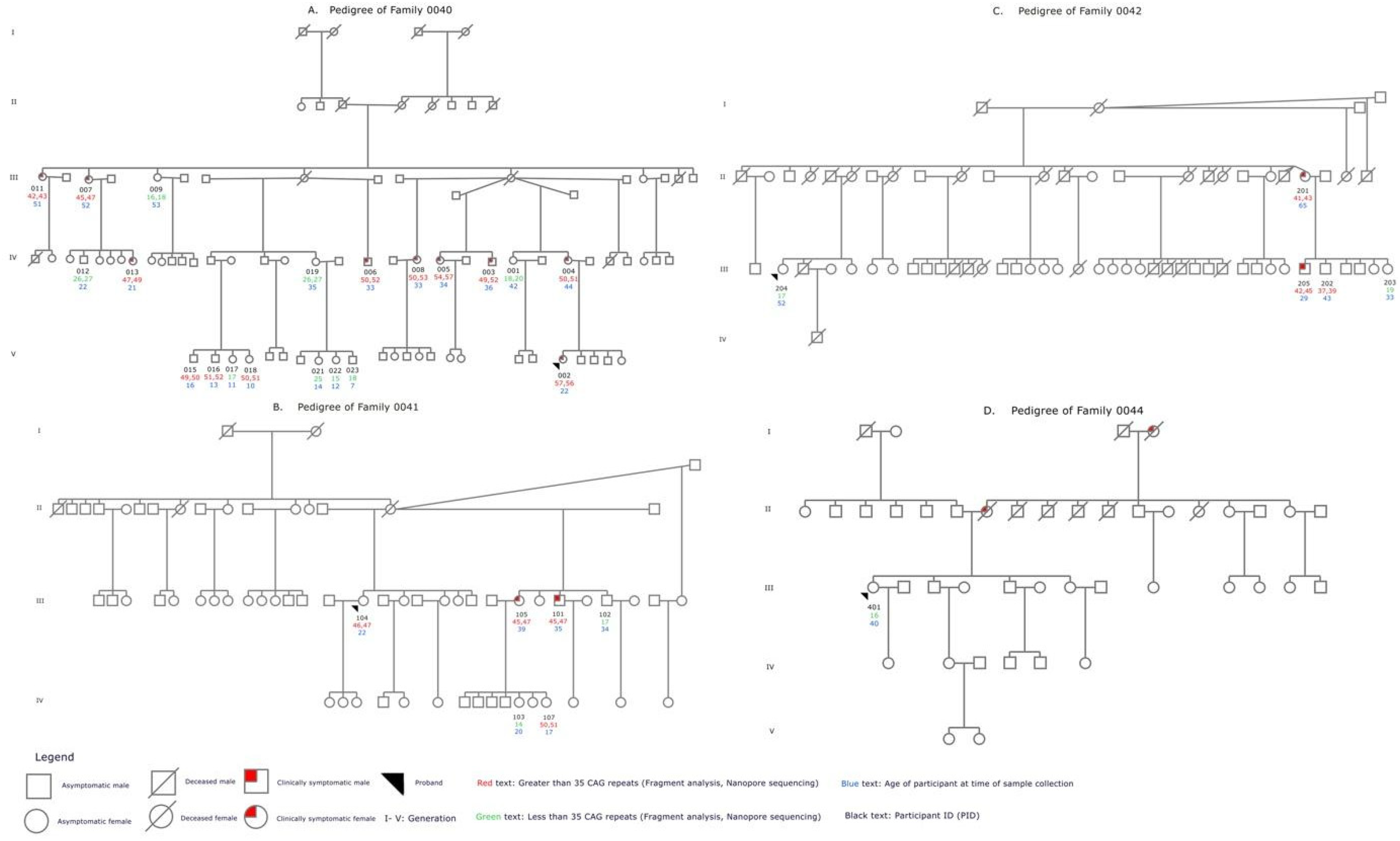
Family Pedigrees summarizing demographic, clinical, and genetic findings. A-D shows the pedigrees for families 0040,0041,0042, and 0044 respectively. Affected family members are shown with red shading. Participant IDs are listed in black text. The number of CAG repeats called for each participant by fragment analysis and nanopore sequencing are shown in red (>35 CAG repeats called) and green (≤ 35 repeats called). The age of the participant at the time of enrolment is shown in blue text.
A total of 20 Afro-Trinidadian participants between the ages of 7–53 years from family 0040 were clinically assessed for HD (Figure 1A). Ten participants were diagnosed as clinically symptomatic for HD; nine of these were ≥22 years and one was a 16-year-old. PIDs 007 and 011 showed impairment in motor, cognitive, behavioral, functional and independence scales, and PIDs 003, 004, 005, 006, 008 and 013 showed impairment in all parameters except for PID 006 who did not show behavioral impairment and refused to participant in the cognitive assessment. PIDs 015 (age 16) and 002 (age 22) showed early clinical symptoms of HD including motor, cognitive and behavioral impairment. The remaining ten participants of this family did not meet the clinical scoring criteria for HD diagnosis and were classified as asymptomatic for the disorder. Four of these were children between 7–14 years, and six were adults between 35–53 years (Figure 1A).
Six Afro-Trinidadian participants between the ages of 17–39 years from family 0041 were clinically assessed for HD (Figure 1B). Two participants were diagnosed as clinically symptomatic for HD; PID 101 (35 years) and 105 (39 years) showed motor, cognitive, and behavioral impairment, and PID 105 also showed functional impairment. Four participants of this family were clinically asymptomatic for HD (Figure 1B).
Five Indo-Trinidadian participants between the ages of 29–65 years from family 0042 were clinically assessed for HD (Figure 1C). Two participants were diagnosed as clinically symptomatic for HD; PIDs 201 (65 years) and 205 (29 years) showed cognitive and behavioral impairment, and PID 201 also showed motor impairment. Three participants of this family were clinically asymptomatic for HD (Figure 1C).
One 40-year-old Indo-Trinidadian participant from family 0044 was clinically assessed for HD and was classified as asymptomatic for the disorder. She had a family history of HD-like symptoms on her mother's side (Figure 1D).
Molecular diagnosis of HD
TP-PCR produced two distinct amplicons of different lengths with an expected faint secondary stuttering pattern on the gel for 19 participants indicating expanded CAG repeats (PIDs 002, 003, 004, 005, 006, 007, 008, 011, 013, 015, 016, 018, 101, 104, 105, 107, 201, 202, 205) (Supplemental Figure 1). One amplicon was observed for 13 participants indicating no expanded CAG repeats (PIDs 001, 009, 012, 019, 107, 021, 022, 023, 102, 103, 203, 204, 401) (Supplemental Figure 1).
Sanger sequencing confirmed that the amplicons obtained by TP-PCR were the CAG repeat tracks of the HTT gene (Supplemental Figure 2). Although this approach gave a minimum estimate of the size of the CAG repeat, it was not possible to assess the exact number of CAG repeats as the sequencing reads did not extend all the way from the unique forward primer sequence to the unique reverse primer sequence.
Fragment analysis and nanopore sequencing found between 6–57 CAG repeats among the study participants. The numbers of CAG repeats in each allele of each participant based on fragment analysis and nanopore sequencing are summarized in Table 1.
Clinical and genetic results for all participants.
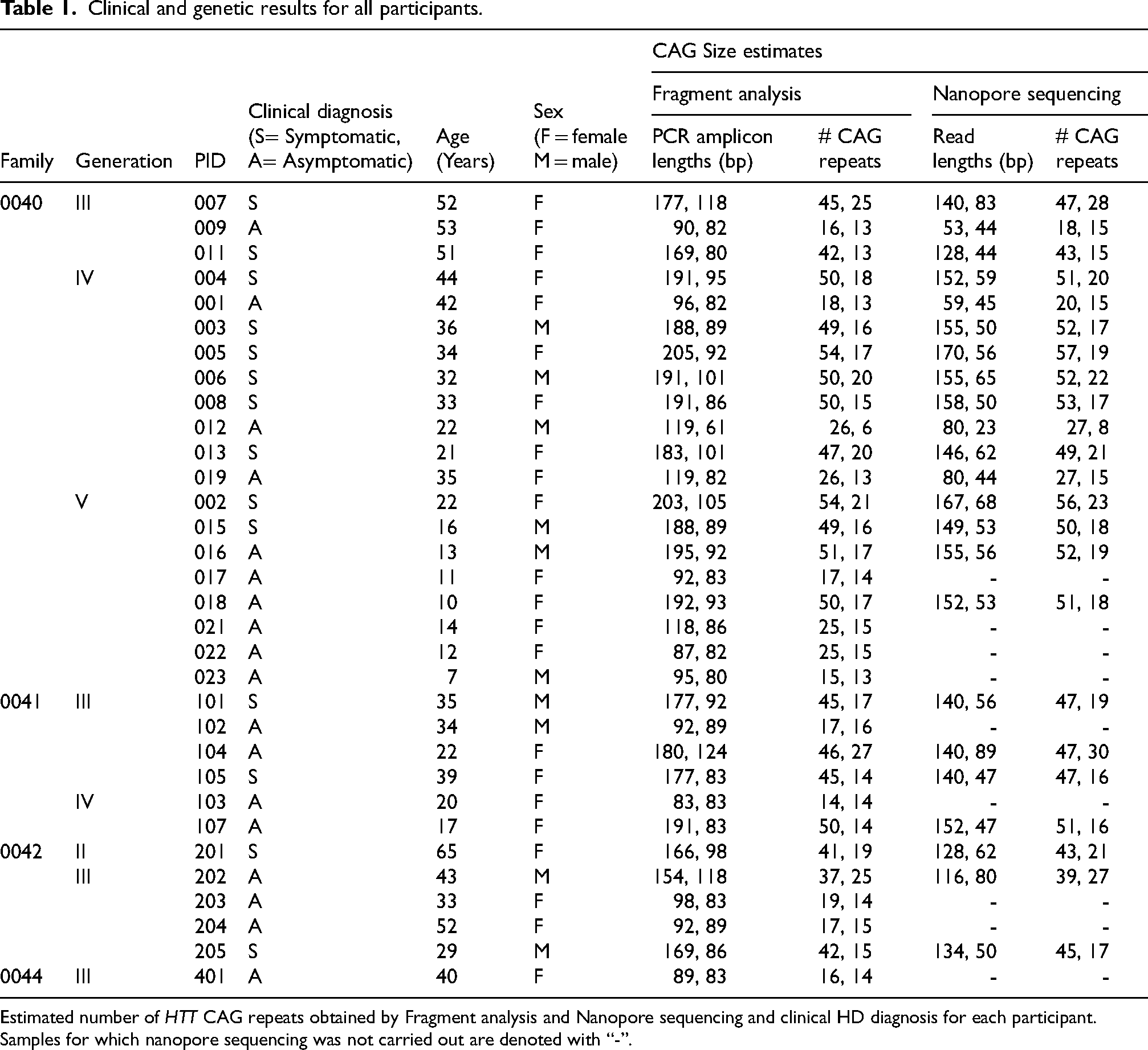
Estimated number of HTT CAG repeats obtained by Fragment analysis and Nanopore sequencing and clinical HD diagnosis for each participant.
Samples for which nanopore sequencing was not carried out are denoted with “-”.
Supplemental Figure 3 shows the fragment analysis electropherograms from which the number of CAG repeats for each participant were derived. Supplemental Figure 4 shows the nanopore read length histograms. The total read counts for Nanopore Sequencing data ranged from 4759–43341 (mean = 21129, median = 19092). Allowing zero mismatches in primer sequence alignment (SW0) resulted in 0.6–5.6% (mean = 2.6%, median = 2.2%) of reads being usable for determining the number of CAG repeats. Increasing the number of mismatches allowed to up to10 (SW10) resulted in 70.4–79.7% (mean = 76.6%, median = 76.7%) of reads being usable.
Genetic and clinical correlations of HD
All 32 participants were successfully genotyped at the HTT CAG locus by fragment analysis and 23 were additionally genotyped by nanopore sequencing of TP-PCR products. Clinical and genetic findings are summarized in Table 1. All symptomatic participants carried pathogenic HTT expansions ranging from 42–57 CAG repeats, confirming their genetic diagnosis of HD. Estimated CAG sizes derived from fragment or read length calculations are presented in Table 1.
A total of 24 adult participants aged 20–65 years were included in the study. The clinical and molecular diagnoses for 22 of the 24 adult participants were fully concordant. These include 13 (four males, nine females) clinically symptomatic participants with between 42–57 CAG repeats and nine (two males, seven females) clinically asymptomatic participants with between 13–27 CAG repeats. The remaining two additional adult participants, ages 22 (female) and 43 (male) years, carried 46–47 and 37–39 CAGs, respectively. They are currently asymptomatic but their genotypes were classified as full penetrance and reduced penetrance, respectively.
Eight individuals <18 years old participated in this study, the clinical and molecular diagnoses for five of these participants were fully concordant. These include one (male) clinically symptomatic participant (49–50 CAG repeats) and four (one male, three females) clinically asymptomatic participants (14–17 CAG repeats). The remaining three currently clinically asymptomatic participants were confirmed to carry expanded (50–52) CAG repeats. These include a pair of siblings (female age 10, male age 13) and an unrelated female 17-year-old. Their genotypes were classified as full penetrance.
Twenty-three participants were genotyped by both fragment analysis and nanopore sequencing (Table 1). These included 14 symptomatic participants with between 41–57 CAG repeats; two asymptomatic adults, one with 46–47 CAG repeats (22 years) and one with 37–39 CAG repeats (43 years, reduced penetrance); four asymptomatic adults with ≤ 27 CAG repeats; and three asymptomatic children with between 50–52 CAG repeats. The total number of CAG repeats for each allele for all 23 participants obtained by fragment analysis (FA) and nanopore sequencing (ONT) methods reveals a strong linear correlation (R2 = 0.998) (Figure 2, left panel]) The best fit line (ONT = 0.997 * FA + 1.70) has an offset of 1.70, signaling either a slight over-reporting in nanopore sequencing or slight under-reporting in fragment analysis (Figure 2, left panel). A Bland-Altman agreement analysis (Figure 2, right panel), indicates a mean difference of 1.62 CAG repeats between the Fragment Analysis and Nanopore Sequencing methods, once again showing that the nanopore sequencing results are reporting slightly higher CAG counts across all categories of HD genotypes. The largest and smallest differences between the ONT and FA were 3 CAG repeats for two samples and 0.33 CAG repeats (1 bp difference) for one sample respectively, and these three participants all carried > 40 CAG repeats. Importantly, the two methods classified each participant into the same category of HD manifestation, despite this small systematic difference in the number of CAGs called by each method. For 17 participants ≥20 years, the genotypes obtained by both approaches were concordant with the clinical phenotypes. Participants for which genotype and clinical phenotype were not concordant are currently clinically asymptomatic and below the expected age of onset.
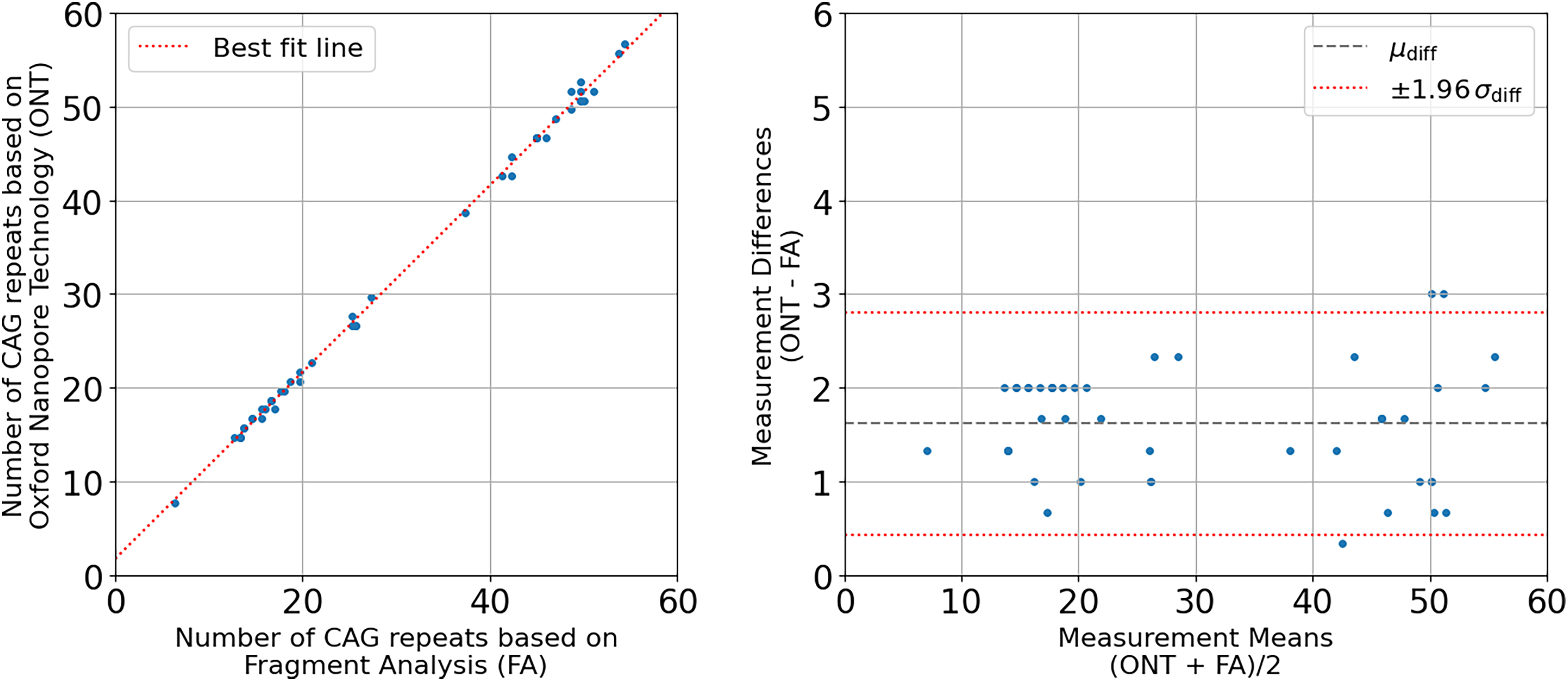
Comparison of number of CAG repeats obtained by nanopore sequencing (ONT) versus fragment analysis (FA). Left Panel: Scatter plot of CAG repeat lengths from 23 participants obtained by nanopore sequencing versus fragment analysis, with a linear regression line overlaid. The regression line represents the best-fit linear relationship between the two variables, with the equation ONT = 0.997*FA + 1.70 and an R2 coefficient of determination of 0.998. Right Panel: Bland-Altman plot showing the agreement between the two methods of measuring the number of CAG repeats, with the mean of the measurements on the x-axis ((ONT + FA)/2) and the difference between the methods on the y-axis (ONT – FA). The mean and standard deviation of the differences are 1.62 and 0.60 respectively. The dashed grey line represents the mean difference, and the red dotted lines represent ±1.96 standard deviations of the differences, corresponding to the 95% limits of agreement for a normal distribution.
Figure 1 shows family pedigrees for each of the four multigenerational families indicating the generation within each family (I-V as appropriate), the clinical HD manifestation of the individual, the number of CAG repeats obtained from both fragment analysis and nanopore sequencing, and the age of each participant at the time of sample collection. In all three families with multiple participants a decreasing age of onset of the disease and progressive expansion of the mutation within each successive generation was observed. In Family 0040, there were 42–47 CAG repeats in participants 51–52 years old in generation III, 47–54 CAG repeats in participants 21–44 years old in generation IV and 49–57 CAG repeats in participants 10–22 years old in generation V. In Family 0041, there were 45–47 CAG repeats in participants 22–39 years old in generation III and 50–51 CAG repeats in a 17-year-old in generation IV. In Family 0042, there were 41–43 CAG repeats in a 65-year-old in generation II and 42–47 CAG repeats in a 29-year-old in generation III. Families 0040 and 0041 had exactly one pair of affected parent and child participants from which we could investigate direct transmission of the expanded allele. In each case, the disease was maternally transmitted and expansion was observed. One 40-year-old participant from Family 0044 did not show any clinical symptoms of the disease and was confirmed to carry 15 CAG repeats, despite a family history of the disease. An association between the CAG expansion of symptomatic participants for HD with each successive generation could not have been assessed because of the lack of additional participation from symptomatic family members in family 0044.
Discussion
In this study we used a TP-PCR assay to generate different sized amplicons from both alleles of each individual representing the CAG repeat region of the HTT gene. We coupled TP-PCR with downstream molecular methods including Sanger sequencing, fragment analysis, and nanopore sequencing with a custom bioinformatics workflow to estimate the size of the HTT CAG repeats in our participants. We confirmed that the amplified allele constituted the HTT CAG repeat region by Sanger sequencing. Fragment analysis and nanopore sequencing binned each participant into exactly the same categories of normal, intermediate, reduced-penetrance, and full-penetrance HD.
Accurate diagnosis of HD relies on both clinical and molecular testing. To our knowledge, this is the first report of genetic confirmation of HD in T&T. We observed expanded CAG repeats in three multigenerational families with symptomatic participants, confirming HD in these families. No expanded CAG repeats were observed in the single asymptomatic participant of family 0044 despite a family history of HD-like symptoms. This may be due to the absence of a canonical HD allele, or an alternative HD–like disorder segregating in family 0044. Progressive expansion of the repeat and decreasing age of onset was observed in each successive generation within each of the three families in which HD mutations were confirmed. Clinical and genetic diagnoses were concordant for most participants. All 14 symptomatic participants (13 adults and one 16-year-old) carried between 42–57 CAGs. Among the 18 asymptomatic participants, 13 carried 13–27 CAGs and are expected to be unaffected by HD. The remaining five currently asymptomatic participants (two adults ages 22 and 43 years, and three children) carry expanded repeats (39–50 CAGs) and are expected to develop symptoms of the disorder in the future due to the fact that across all observed populations, expanded repeats inevitably result in clinical onset of HD.
Although TP-PCR predominantly amplified the full length alleles, the reverse primer sequence contained multiple CTG triplet bases which introduced non-specific DNA amplification by random annealing within the CAG repeat track of the target gene. This resulted in the expected stuttering pattern in the visualization of the PCR amplicons by gel electrophoresis, the low frequency stuttered peaks in the electropherograms, and the low frequency stuttered peaks in the read length histograms from the nanopore data. The reverse primer also spans the canonical intervening sequence (CAA CAG CCG CCA). Therefore, this approach is expected to selectively amplify canonical alleles and provides no information on the nature of the HTT intervening sequence.
Using the electropherograms generated from fragment analysis, the length of each PCR fragment was estimated from the most frequent peak/s of the stuttered pattern. The size of the allele was not considered an absolute value for calculating the number of CAG repeats because of the possibility of instrument variability and the fluorescent molecule slightly changing the migration of the fragment compared to the size standard. The estimated length of each PCR fragment can be confidently converted to an absolute number of CAG repeats by comparison to a control sample with a known number of CAG repeats. However, no such control was available at the time of data analysis.
We attempted to use the most frequent read lengths obtained from TP-PCR products sequenced with ONT as a proxy to count the exact numbers of CAG repeats in the two alleles of each sample. Primer sequence alignment using Smith-Waterman with zero mismatches (SW0) resulted in only 0.6–5.6% of the reads being used to generate histograms indicating that the ONT sequences contained a high number of base-calling errors in the primer sequences. This low number of reads made it difficult to determine the most frequent read lengths because there were no prominent peaks in the histograms. To include a larger percentage of reads with the expectation of generating histograms with at least two read lengths of significantly high frequencies, Smith-Waterman with up to ten mismatches (SW10) was run at the expense of reducing the quality and accuracy of the bases. Similar histograms were generated but with slightly different most frequent read lengths compared to SW0. As such, we were only able to achieve an approximate determination of the number of CAG repeats and not an exact count using ONT. The systematic difference between the number of CAGs obtained by the two methods may be an artefact of the low number of usable reads for the nanopore bioinformatics workflow, or be due to the inaccurate determination of CAG length from the fragment analysis data lacking a control. Increasing the read depth and inclusion of a control may provide further insight into the observed difference. Additionally, the progress in Nanopore technology (V14) for heightened accuracy, has the ability to further increase application of the described method.
Due to these limitations, both TP-PCR with fragment analysis and nanopore sequencing were unable to confirm absolute lengths of the CAG expansion. Nonetheless, both methods produced slightly different, albeit highly correlated (R2 = 0.998), estimates of the number of CAG repeats. Further genotyping would be needed to accurately determine CAG size for clinical diagnoses. For the individuals in this study, improved genotyping accuracy may help clarify the clinical prognosis of the 43-year-old asymptomatic individual with 37–39 repeats, currently characterized as carrying an incompletely penetrant allele.
The development of novel gene-based therapeutics with the potential to transform the clinical management of monogenic disorders such as HD is imminent. But, equitable global implementation of such technologies will rely on equitable access to accurate genetic diagnosis of the disease in all regions. Capacity building efforts such as the Human Health Heredity and the Environment in Africa (H3Africa) project have started to close this access gap, and recently reported a genetically confirmed Malian HD cohort with 18 participants, one of the largest across Africa. 28 The COVID-19 pandemic further accelerated the adoption of molecular diagnostics in the Caribbean and other lesser resourced regions. 29 So, genetic diagnosis of HD and other genetic disorders should become more routine in the near future, particularly in traditionally under-resourced settings. Nonetheless, these findings add to the limited data describing HD in the Caribbean region, which has previously been underrepresented in the global HD landscape. Importantly, HTT CAG expanded repeats were found to explain HD cases in both major subpopulations of T&T; two unrelated families of West African ancestry and a family of Indian ancestry. Thus, it is likely that the HD alleles in the different families of our study are of independent origin. This work demonstrates that Caribbean HD cases, regardless of ancestry, may be eligible for inclusion in future global HD therapeutic development and clinical trials.
Footnotes
Acknowledgements
The authors would like to acknowledge the study participants for their consent and trust in the research team. We would also like to acknowledge Sheherazade Abrahim, Akili Joseph, and Neeta Oudit for phlebotomy support. We also thank Christine Carrington and members of her Molecular Genetics and Virology team in the UWI Department of Preclinical Sciences for in-kind contributions to nanopore sequencing and associated bioinformatic analyses, and acknowledge the roles of their funders (Pan American Health Organisation / World Health Organisation, UK Health Security Agency, AHF Global Public Health Institute, and Trinidad and Tobago Ministry of Health) in developing the genomic sequencing capacity used for this project. Finally, we acknowledge Yugesh Ramkhelawan for support with graphics.
ORCID iDs
Ethical considerations
This study was approved by the Campus Research Ethics Committee of The University of the West Indies (UWI), St Augustine, Trinidad on July 29, 2019 (CEC1176/06/09) and by the Eastern Regional Health Authority, Trinidad and Tobago (ERHA-REC:027/08/2019).
Consent to participate
All participants provided written informed consent prior to enrolment in the study. This research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki.
Consent for publication
Written informed consent was provided by the participant(s) or a legally authorized representative for anonymized patient information to be published in this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received no external funding. We acknowledge funding from the MSc in Biotechnology programme at the Department of Life Sciences, The University of the West Indies, St. Augustine, Trinidad.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The Sanger sequencing, fragment analysis, and processed nanopore sequencing data supporting the findings of this study are available within the Supplemental Material. The raw nanopore sequencing data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Supplemental material
Supplemental material for this article is available online.