
Abstract
Progesterone (P4) maintains uterine quiescence during the majority of pregnancy, whereas diminished progesterone receptor (PR) expression and/or activity (ie, functional P4 withdrawal) promotes parturition. To investigate the regulation of PR expression in cervical stroma, fibroblasts from premenopausal hysterectomy specimens were prepared. Greater than 99% of the cultures were vimentin positive (mesenchymal cell marker) with only occasional cytokeratin-8 positivity (epithelial cell marker) and no evidence of CD31-positive (endothelial cell marker) cells. Cells were immunolabeled with antibodies directed against PRs (PR-A and PR-B), estrogen receptor α (ER-α), and glucocorticoid receptor-α/β (GR-α/β). All cells were uniformly immunopositive for ER-α and GR-α/β but did not express PRs. Incubation of cells with 10−8 mol/L 17β-estradiol induced a time-dependent increase in PR-A and PR-B messenger RNAs (mRNAs) by quantitative real-time polymerase chain reactions and proteins by immunoblotting and immunofluorescence. Incubation of cervical fibroblasts with PR ligands (medroxyprogesterone acetate or Org-2058) downregulated PR-A and PR-B levels. Coincubation of cells with PR ligands plus RU-486, a PR antagonist, partially abrogated agonist-induced receptor downregulation. Dexamethasone, a pure glucocorticoid, had no inhibitory effect on PR expression. These results indicate that progestins and estrogens regulate PR expression in cervical fibroblasts. We postulate that hormonal regulation of PR expression in the cervical stroma may contribute to functional P4 withdrawal in preparation for parturition.
Introduction
In all mammalian species studied to date, progesterone (P4) promotes uterine quiescence during most of pregnancy when the myometrium is poorly contractile and the cervix is elongated and closed. 1 -4 This has led to the long-held notion that P4 is the principal block of myriad environmental triggers that would otherwise drive the emptying of the uterus. 2,5,6 In addition, P4 acts locally on the uterine cervix to prevent premature remodeling. 7
Recently developed in vivo and in vitro model systems have permitted investigation of progesterone receptor (PR) functions in the context of parturition in myometrial, decidual, and cervical tissues. 8 -14 Mifepristone (RU486) administration in a mouse model revealed that disruption of PR signaling elicited preterm birth. 15 -17 Moreover, clinical trials carried out in the last few years indicated that systemic administration of 17α-hydroxyprogesterone caproate or vaginal instillation of P4 reduces the incidence of preterm birth in singleton pregnancies and in women with a prior history of preterm birth or a short cervix, respectively. 18 -20 Thus, targeting the PR signaling pathway has reemerged as a potentially powerful pharmacologic tool to prevent preterm birth in some at-risk populations. 18,19,21 -23
The PRs are members of the nuclear hormone receptor superfamily (subfamily 3, NR3C3) present in female reproductive tissues, including endometrium/decidua, myometrium, cervix, and breast. 8,24 -29 To examine the role of PR function and regulation in the uterine cervix, we developed a primary culture model using cervical stromal cells to study regulation of PR expression in cervical fibroblasts. The 2 canonical PR isoforms, that is, PR-A (96 kDa) and PR-B (116 kDa), are expressed in cervical stromal fibroblasts after pretreatment with 17β-estradiol (17β-E2) but are virtually undetectable without estrogen priming. Our experiments demonstrate that ligation of PRs with progestin agonists downregulate both PR-A and PR-B isoforms following 17β-E2 priming. Moreover, we show that dexamethasone (Dex), a potent and selective glucocorticoid ligand, does not alter 17β-E2–induced PR levels, indicating that progestin agonists specifically regulate PRs. The regulation of PR expression may be one mechanism by which P4 governs the maintenance of cervical integrity during the bulk of pregnancy. These studies provide the first evidence that progestins can downregulate PR expression in cervical stromal fibroblasts.
Methods
Cell Culture and Validation
Following institutional review board approval for tissue collections, deidentified cervical tissues were obtained from premenopausal women undergoing hysterectomy for benign gynecological conditions through the Cooperative Human Tissue Network of the Ohio State Wexner Medical Center. Primary cultures of human cervical fibroblasts were established from explants of fragments (2-3 mm3) of cervical stroma obtained from dissecting the ecto- and endocervix and seeding both tissues onto tissue culture plates sandwiched beneath glass coverslips. The cells were grown in complete basal medium consisting of high-glucose (4.5 g/L) Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 50 μg/mL gentamicin sulfate, and 0.5 μg/mL amphotericin B. Cultures were replated and grown to 80% to 90% confluence, and media were replenished at 2-day intervals. Cultures were tested for mycoplasma contamination every 6 months using the MycoAlert Mycoplasma Detection Kit (Lonza, Basal, Switzerland). Enrichment of cervical fibroblasts in the resulting monolayer cultures was confirmed by immunofluorescence (IF) staining for vimentin, cytokeratin 8 (CK-8), and CD31 (see Immunofluorescence section subsequently). Cervical cells from 3 separate tissue donors were used for the studies described, with experiments performed between the third and seventh passages.
Immunohistochemistry
Portions of the cervical tissues used to establish cultures were fixed with 10% neutral-buffered formalin for 24 hours, dehydrated in a graded ethanol series to xylene, and finally embedded in paraffin. Sections (5-6 µm) were cut and placed onto clean glass microscope slides. After deparaffinizing with xylene and rehydrating through a graded ethanol series, antigen retrieval was accomplished by incubating sections in 10 mmol/L sodium citrate (pH 6.0) with 0.05% Tween-20 in an autoclave (4 minutes at 18 psi/122°C). Sections were then labeled using mouse monoclonal anti-human PR (Clone PgR 1294, Dako, 1:100, Capinteria, CA) and the UltraVision LP Detection System (ThermoFisher Scientific, Waltham, MA), according to the manufacturer’s specifications. The sections were counterstained with Mayer’s hematoxylin (Sigma-Aldrich, St. Louis, MO), dehydrated with ethanol and xylene, and then mounted on coverslips using Permount (Fisher Scientific, Pittsburgh, PA). To assess primary antibody specificity, serial sections were treated identically, but with an equivalent concentration of normal mouse immunoglobulin (Ig) G1 isotype control (Santa Cruz Biotechnology, Dallas, TX) was substituted for the primary antibody. Digital images were captured using a Nikon Eclipse 90i microscope equipped with a SPOT RT3 CCD (Diagnostic Instruments, Sterling Heights, MI) operated in the color imaging mode.
Cell Treatments
For immunofluorescence microscopy, cervical fibroblasts were seeded (5 × 104/well) onto 12-mm glass coverslips inserted into 24-well plates in complete medium. For RNA or nuclear protein extractions, cells were plated into 100 mm dishes at a density of 106/dish in complete medium. After 2 days, media were changed to medium composed of phenol red-free DMEM/F12 (1:1) supplemented with 0.5% charcoal-stripped FBS. In time course experiments, cells were stimulated in the absence (vehicle control) or presence of 17β-estradiol (17β-E2, 10−8 mol/L) for 3 to 12 days. For end point experiments, cells exposed to 17β-E2 or solvent control were also incubated in the presence or absence of medroxyprogesterone acetate (MPA, PR agonist and weak glucocorticoid receptor [GR] and androgen receptor [AR] agonist, 10−7 mol/L, Sigma-Aldrich [St. Louis, MO]), Org-2058 (highly selective PR agonist, 10−7 mol/L, Organon [Roseland, NJ]) or Dex (highly selective GR agonist, 10−8 mol/L, Sigma-Aldrich [St. Louis, MO]) for 7 days. In some experiments, RU-486 (mixed PR/GR antagonist, 10−6 mol/L, Sigma-Aldrich [St. Louis, MO]) was coincubated together with MPA or Org-2058 during the 7-day incubation period. Control cells were exposed to 0.01% ethanol vehicle.
Immunofluorescence Microscopy
Cultures treated as described earlier in addition to cells fixed in the absence of treatments (where described earlier) were fixed with 4% paraformaldehyde/phosphate-buffered saline (PBS) for 30 minutes and then washed with PBS. Cells were permeabilized with 0.2% Triton X-100/PBS for 15 minutes, followed by additional rinsing with PBS, and then blocking with 5% normal goat serum/PBS for 60 minutes. Coverslips were incubated with primary antibodies in blocking solution overnight at 4°C. The primary antibodies used were mouse monoclonal anti-human PR (Clone PgR 1294, 1:100, Dako, Capinteria, CA), rabbit polyclonal antiestrogen receptor alpha (ERα; ab37438, 1:200, Abcam, Cambridge, MA), rabbit anti-human GR-α/β (1:100, R&D Systems), mouse monoclonal anti-vimentin (clone V9, 1:200, Sigma-Aldrich, St. Louis, MO), rabbit polyclonal anticytokeratin 8 (1:200, Sigma-Aldrich, St. Louis, MO), and mouse anti-CD31 (Clone 9G11, 1:100, R&D Systems, Minneapolis, MN). Following additional rinses with blocking solution, coverslips were incubated with secondary antibodies conjugated either to AlexaFluor-488 or AlexaFluor-594 (Life Technologies, Grand Island, NY), followed by rinsing and treatment with Prolong antifading agent containing 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies, Grand Island, NY).
Digital 14-bit epifluorescence images were collected with a Nikon Eclipse 90i microscope equipped with a SPOT RT3 CCD (Diagnostic Instruments, Sterling Heights, MI) operated in the monochrome capture mode. Five to 10 randomly selected, nonoverlapping fields from each treatment group in each experiment were captured using the SPOT Advanced version 4.7 imaging software (Diagnostic Instruments, Sterling Heights, MI). Acquisition parameters were held constant throughout imaging, and the absence of pixel saturation was confirmed for each image. Measurements for each comparison group were performed together to minimize variation in excitation illumination. Fluorescence intensity measurements were performed using images imported into Slidebook version 5.0 (Intelligent Imaging Innovations, Fremont, CA). Annotations for antibodies used for immunohistochemistry and IF studies are located in Supplemental Table 1.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction Analysis
Total RNA was extracted using TRIzol reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s protocol up to the chloroform extraction and centrifugation step. The resulting aqueous phase was mixed with an equal volume of 70% ethanol, applied to an RNeasy mini-column (Qiagen, Valencia, CA) and processed per the manufacturer’s recommendations with on-column DNase digestion. Total RNA was quantified by UV absorbance at 260 and 280 nm on a NanoDrop 2000 (Thermo Scientific, Waltham, MA), and 2 μg of total RNA from each sample was reverse transcribed to complementary DNA (cDNA) using random hexamers with SuperScript III Reverse Transcriptase (Life Technologies, Grand Island, NY). Quantitative RT-PCR was performed using an equal amount of cDNA per sample on a LightCycler 480 II System (Roche Applied Science, Indianapolis, IN) using primer/probe sets specific to PR-B and PR-A/B (Roche Applied Science, Indianapolis, IN) and Large Ribosomal Protein (RPLP0) RNA (Applied Biosystems, Grand Island, NY) with the LightCycler 480 Probes Master Mix. The results were analyzed with the LightCycler 480 software. See Supplemental Tables 2 and 3 for primer sequences and thermal cycling conditions, respectively.
Protein Extraction and Immunoblot Analysis
Nuclear proteins were extracted using a nuclear extract kit (Active Motif, Carlsbad, CA), according to the manufacturer’s instructions. Briefly, cells were rinsed in PBS, collected by centrifugation, then resuspended in a hypotonic lysis buffer containing protease inhibitors, and incubated for 15 minutes on ice. Nonionic detergent NP-40 was added to a final concentration of 0.5% (v/v), and the cells were subjected to vigorous vortexing. Nuclear pellets were collected by centrifugation at 14 000 × g/4°C for 30 seconds and resuspended in 50 µL of complete lysis buffer (Active Motif, Carlsbad, CA, proprietary formulation) containing freshly added 1 mmol/L dithiothreitol, then incubated on ice for 30 minutes with rocking. Finally, nuclear extracts were clarified by centrifugation at 14 000 × g/4°C for 10 minutes, and protein concentration determined using a detergent compatible colorimetric assay kit (Bio-Rad, Hercules, CA). After quantifying protein concentration using bovine serum albumin as a standard, 10 μg per sample was fractionated using Novex Tris–glycine 10% acrylamide gels (Life Technologies, Grand Island, NY). Proteins were then transferred to nitrocellulose membranes using either traditional wet transfer methods or the iBlot dry blotting system (Life Technologies, Grand Island, NY). After blocking with 5% nonfat dry milk/Tris-buffered saline and 0.2% Tween-20 (pH 7.8), membranes were probed with mouse anti-human PR (1:1000, Dako, Capinteria, CA) by rabbit antimouse IgG-horseradish peroxidase (1:2000, Thermo Scientific, Pittsburgh, PA). Antibody binding was detected using SuperSignal chemiluminescent substrate (Thermo Scientific, Pittsburgh, PA) in concert with a VersaDoc Imaging System (Bio-Rad, Hercules, CA). Following detection, blots were reprobed using a monoclonal anti-TATA-binding protein (TBP; 1:1000, clone 58C9, Sigma-Aldrich, St. Louis, MO) as a control for protein loading; all remaining incubation steps were identical to the original blotting procedure. All chemiluminescent signals were captured using the VersaDoc Imaging System (Bio-Rad, Hercules, CA) using settings within the linear range of the CCD camera.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism version 5.01 (GraphPad Software, La Jolla, CA). D’Agostino and Pearson omnibus normality testing performed on fluorescence intensity data revealed they were not normally distributed. Consequently, the data were analyzed for statistical significance by the method of Kruskal–Wallis followed by post hoc testing by the method of Dunn and expressed as mean ± standard error of the mean unless otherwise noted.
Results
Validation of Cervical Stromal Fibroblast Cultures
Primary cultures were established from nonmalignant cervices obtained at the time of hysterectomy in premenopausal women for clinical indications not involving the cervix. Small fragments of cervical tissue were dissected from overlying epithelium and cultured as explants (Figure 1A). After 21 to 25 days, outgrowths comprised primarily of largely stromal fibroblasts grew out into a nearly confluent monolayer (Figure 1B).
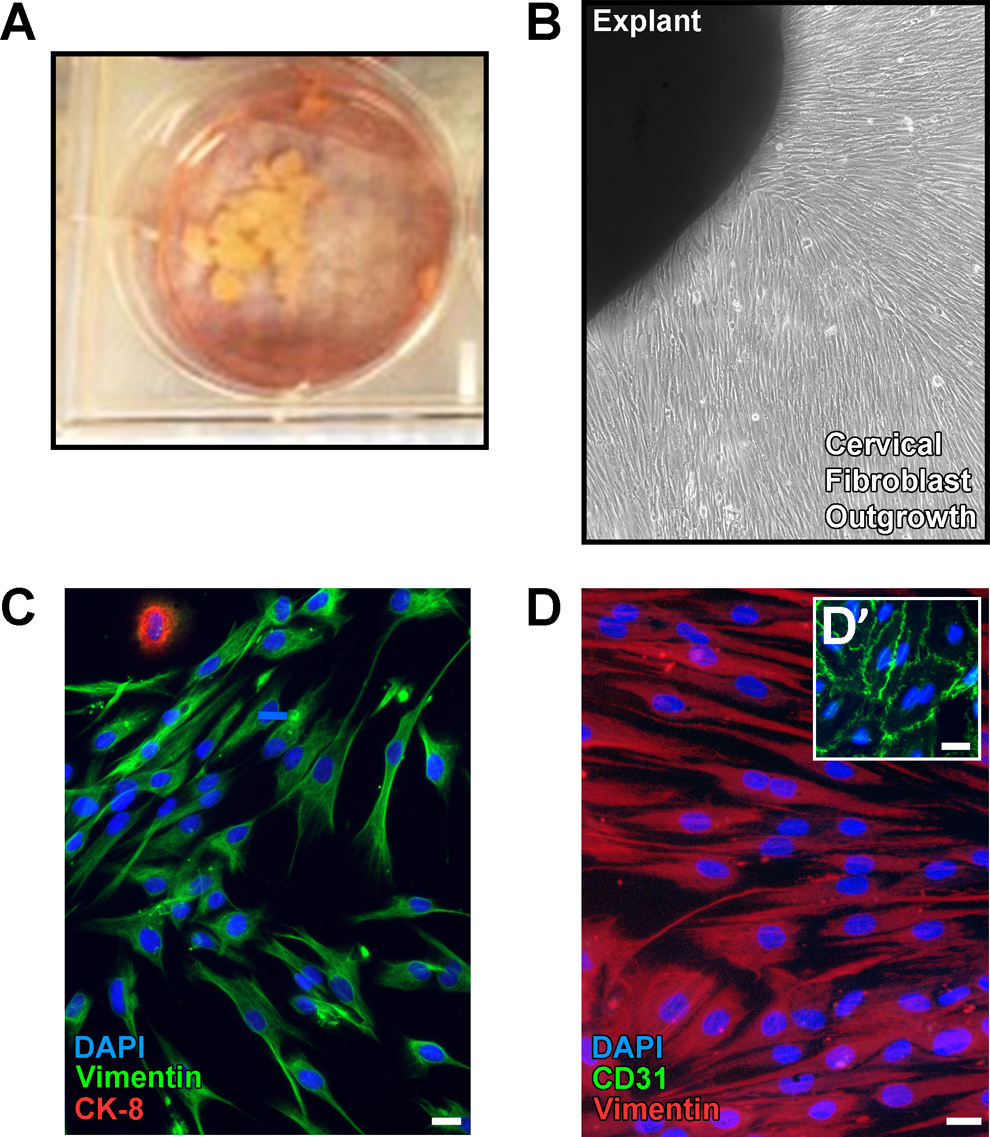
Establishment and initial characterization of cervical fibroblast cell cultures. A, Photograph depicting small fragments of explanted cervical stromal tissue held in place under glass coverslips and grown in individual wells of a 6-well tissue culture plate in complete medium. B, Low magnification phase contrast image of cellular outgrowth from explanted cervical tissue after 21 days in culture. C, Representative immunofluorescence image of cervical fibroblasts grown under basal conditions (untreated) and coimmunolabeled using antibodies directed against vimentin (green) and cytokeratin 8 (CK-8, red), followed by counterstaining of nuclei with 4′,6-diamidino-2-phenylindole (DAPI; blue). By the second passage following the initial outgrowth period, >99% of the cells demonstrated vimentin labeling (marker for mesenchymally-derived cells), whereas only an occasional cell labeled with anti-CK8 (epithelial marker). Scale bars = 20 μm. D, Cells cultivated as in panel C were coimmunolabeled using antibodies detecting CD31 (green, endothelial marker) and vimentin (red) with DAPI counterstaining. CD31 labeling was absent in cervical cells but was readily detected in human umbilical vein endothelial cells (green in D’). Scale bars = 20 μm. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
Replating cells onto glass coverslips after the initial explant culture period and immunofluorescent staining for fibroblast, epithelial, and endothelial markers revealed that cultures were >99% immunoreactive for vimentin with only occasional cytokeratin-immunoreactive cells and no evidence of endothelial cells, leukocytes, or smooth muscle cells (Figure 1C and D). After the second passage, 100% of cells in culture were immunopositive for vimentin. These observations confirmed that our culture method selectively enriches for stromal fibroblasts with minimal to no sustained contamination from surface and glandular epithelial cells or endothelial cells. Mycoplasma testing determined that the cultures are negative for infection.
Stromal Fibroblasts Express PRs Following 17β-E2 Priming
Tissue sections of cervix from nonpregnant, premenopausal specimens containing stroma and epithelial components were stained with a monoclonal antibody directed against total PR (PR-A and PR-B). The PR immunoreactivity was detected in the basal layer of epithelium and throughout the stroma in all tissue sections examined (Figure 2).
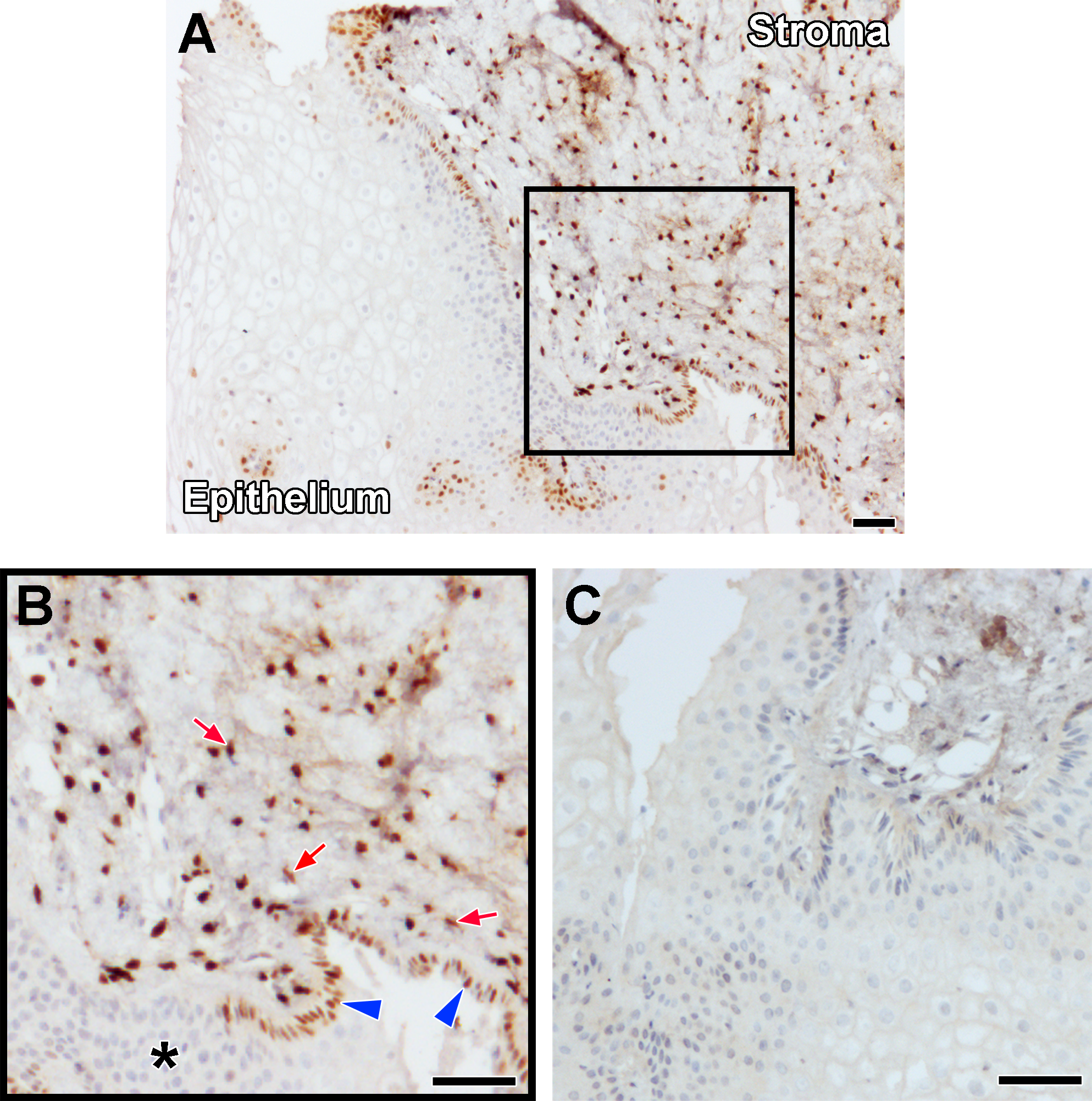
Progesterone receptor (PR) expression in premenopausal cervical biopsy specimens. A, Histological section of cervical tissue immunolabeled with an antibody directed against PR (brown). B, Detail of area within box in panel A. Red arrows indicate PR immunolabeling in nuclei of cervical stromal fibroblasts, while blue arrowheads denote PR expression in basal squamous epithelial cells of the ectocervix. PR labeling was also observed in columnar epithelial cells of the endocervix and glands but was undetected in the more superficial layers of the ectocervix (asterisk). C, Serial section of the biopsy shown in previous panels immunolabeled with an isotype control antibody at a concentration equivalent to the anti-PR antibody. Scale bars = 100 μm. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
Under basal culture conditions, passaged cervical fibroblasts expressed ERα constitutively (Figure 3A), but unlike the parental cells in vivo, lacked robust PR expression. In addition, all stromal fibroblasts were immunoreactive with anti-GR antibodies (Figure 3B). Based upon the human menstrual cycle 27,30 in which endometrial PRs are present at high levels only after estrogen priming, we treated cervical stromal cells with physiological concentrations of 17β-E2 (10−8 mol/L) for 3 to 12 days and then tested for the presence of PR expression. Using quantitative real-time polymerase chain reaction (qRT-PCR) analysis of stromal cell cultures exposed to 17-βE2, we detected a time-dependent increase in total PR (ie, the region of the transcript common to both isoforms) and PR-B messenger RNA (mRNA) expression (Figure 3C). These observations were confirmed by immunofluorescence using a monoclonal antibody that recognizes both PR isoforms (Figure 4D). 31 Finally, Western blotting also confirmed the in vitro expression of PR-A and PR-B in the presence of 17β-E2 (Figure 4E).
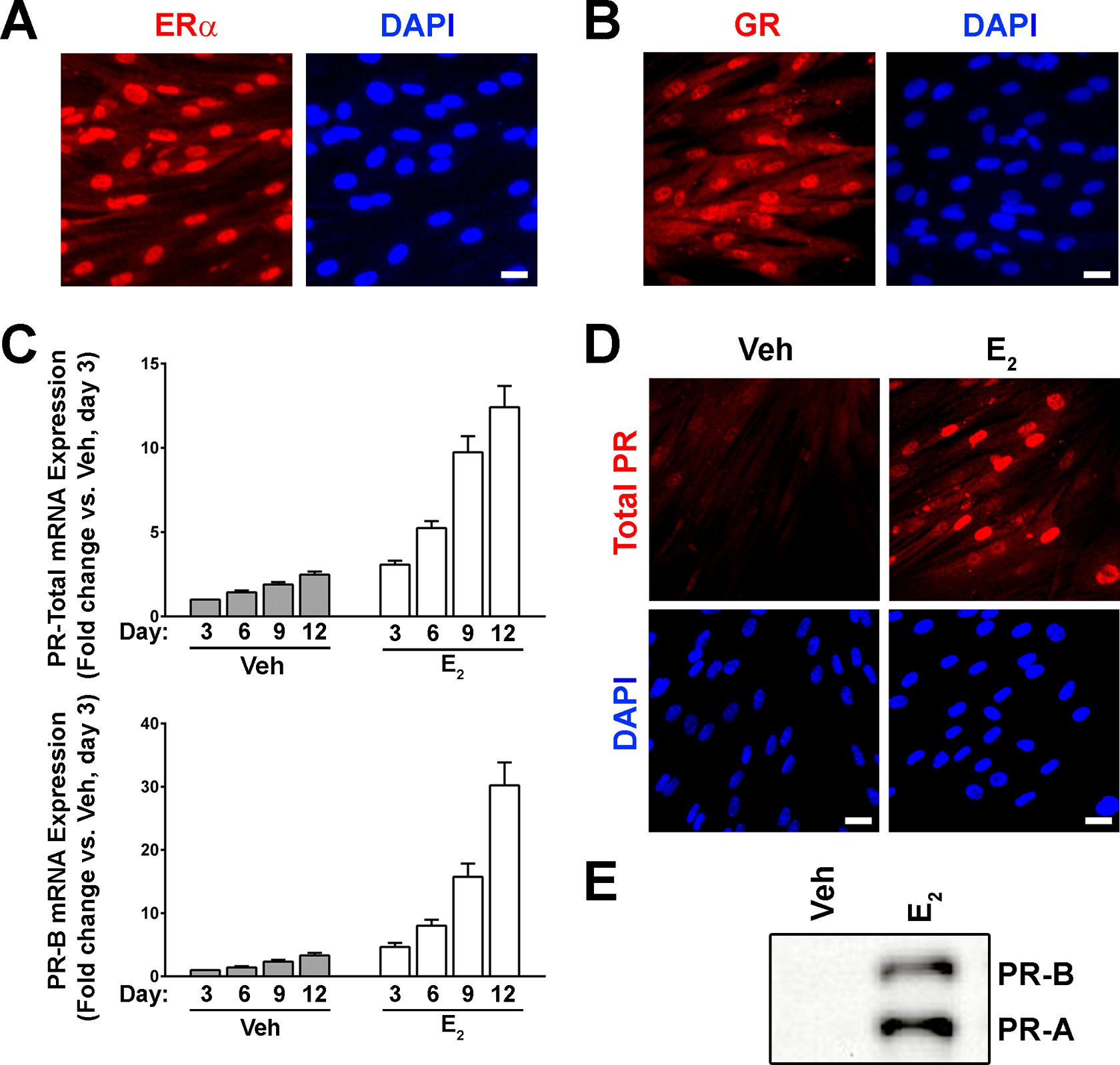
Cervical fibroblasts cultured in vitro constitutively express estrogen receptor α (ERα) and glucocorticoid receptors α/β (GR-α/β), whereas progesterone receptor (PR) expression is inducible following 17β-estradiol (17β-E2) priming. A, Immunofluorescence images demonstrating ERα and (B) GR immunolabeling (red) and DAPI-stained nuclei (blue) in cervical fibroblasts grown under basal conditions. Scale bar = 20 μm. C, Cervical fibroblasts were incubated in treatment medium (see Materials and Methods section) for 3 to 12 days containing either vehicle alone (Veh, 0.01% ethanol, gray bars) and/or 10−8 mol/L 17β-E2 (white bars), and PR messenger RNA (mRNA) expression was measured by quantitative real-time polymerase chain reaction (qRT-PCR) using primer/probe sets detecting total PR (through amplification of the transcript region that is common to both PR-A and PR-B) or PR-B only. Samples were normalized to RPLP0 mRNA and expressed as the fold-change relative to Veh-treated cells collected after 3 days of treatment (mean ± standard error of the mean [SEM], 2 independent experiments). D, Representative immunofluorescence images demonstrating induction of nuclear PR expression (red) following a 7-day incubation in 17β-E2. Scale bars = 20 μm. E, Nuclear extracts prepared from cervical fibroblasts following incubation in the absence or presence of 17β-E2 for 7 days were analyzed by immunoblotting using an antibody detecting PR. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
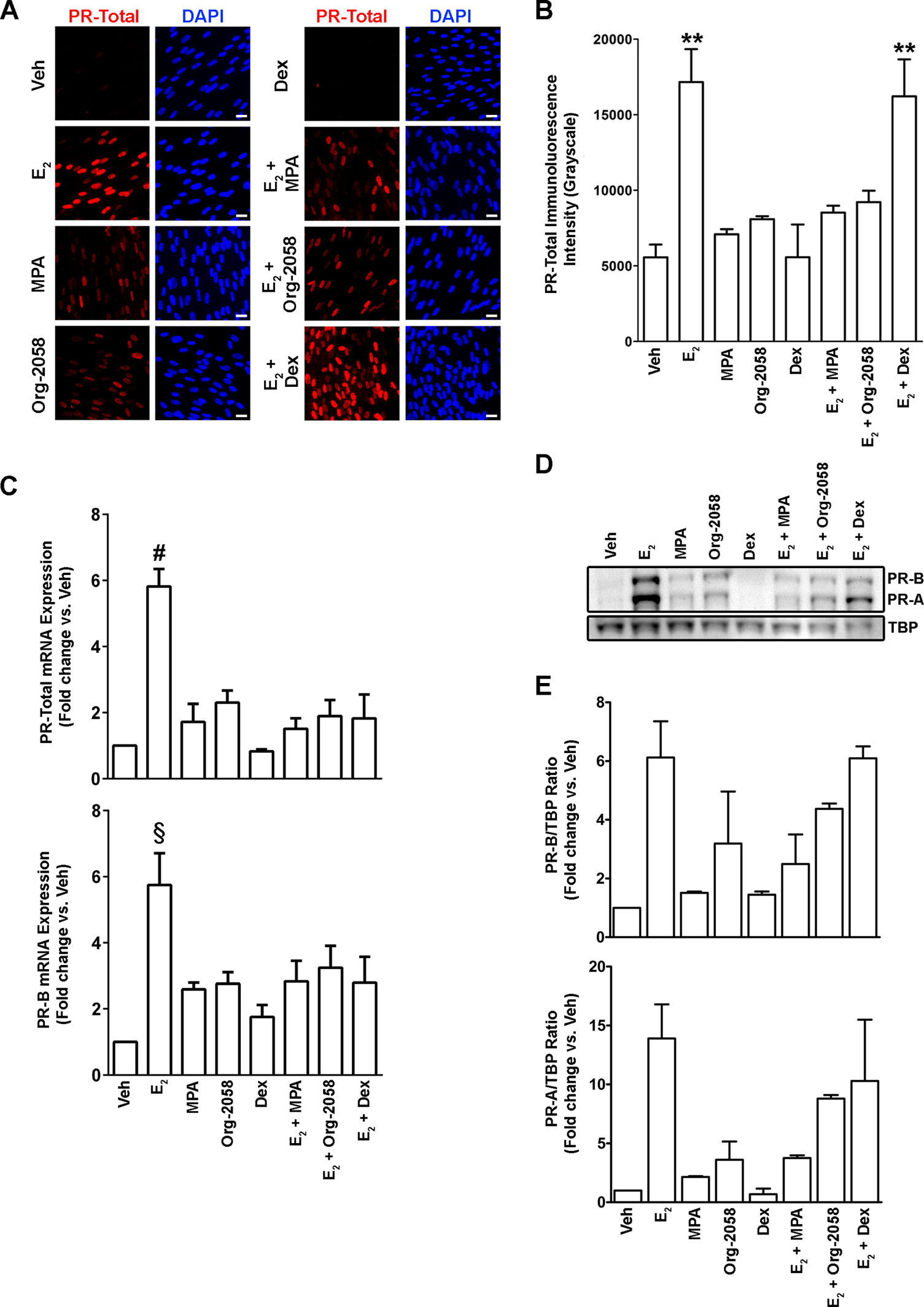
Effects of PR and GR agonists on PR expression. Cervical fibroblasts were incubated in medium with or without 10−8 mol/L 17β-estradiol (17β-E2) in the absence or presence of medroxyprogesterone acetate (MPA, 10−8 mol/L), Org-2058 (10−8 mol/L), or Dex (10−8 mol/L) for 7 days. A, Representative immunofluorescence images demonstrating nuclear PR expression (red) and the number of nuclei per field (DAPI, blue) in each of the treatment conditions. Scale bars = 20 μm. B, Bar graphs (mean ± SEM) summarizing measurements of PR-like fluorescence intensity in 3 independent experiments (**P < .01 vs Veh; Kruskal–Wallis test with Dunn’s multiple comparison posttest). C, qRT-PCR analysis of PR-Total and PR-B mRNA expression. Samples were normalized to RPLP0 mRNA and expressed as the fold-change relative to Veh-treated cells. Bars represent mean ± SEM from 3 independent experiments (# P < .01 vs Dex; § P < .01 vs Veh; Kruskal–Wallis test with Dunn’s multiple comparison posttest). D, Representative immunoblot (from 3 separate experiments) demonstrating PR-A and PR-B protein expression in cervical fibroblasts following 7 days in each treatment condition; as a loading control, blots were reprobed using an antibody against TATA-binding protein (TBP). E, Densitometric analysis of PR-A and PR-B immunoblots, normalized to TBP and expressed as fold change relative to Veh (mean ± SD, 2 independent experiments). PR indicates progesterone receptor; GR, glucocorticoid receptor; DAPI, 4′,6-diamidino-2-phenylindole; SEM, standard error of the mean; qRT-PCR, quantitative real-time polymerase chain reaction; mRNA, messenger RNA; SD, standard deviation. (The color version of this figure is available in the online version at http://rs.sagepub.com/.)
Progesterone receptor Agonists Downregulate PR Expression
The metabolically stable PR agonist MPA is routinely used in studies of PR function. 11,32 However, MPA has both strong PR and weak GR and AR agonist properties, rendering it incompletely PR selective. 33,34 Prior studies by the Lockwood laboratory demonstrated that in decidual cells, MPA downregulates PR-A and PR-B protein levels in the presence of 17β-E2. 11,28 To test whether MPA also downregulates PRs in cervical stromal fibroblasts, we grew cells in the presence or absence of 17β-E2 with or without MPA (10−7 mol/L) for 7 days, followed by immunofluorescence and immunoblot analysis for PR expression. As expected, treatment of cells with 17β-E2 dramatically upregulated PR protein expression, whereas immunofluorescence revealed that MPA exposure reversed 17β-E2–induced PR expression to near background levels.
To determine whether the effects of MPA on PR expression resulted from binding to the PR rather than to the GRs, experiments were expanded by using the highly selective PR agonist Org-2058 and GR agonist Dex in combination with 17β-E2. IFM revealed that exposure of 17β-E2–primed cervical fibroblasts to Org-2058, like MPA, caused downregulation of PR-A and PR-B. In contrast, Dex had no demonstrable effect on PR expression either in the absence or presence of 17β-E2 (Figure 5A and B). Using the same treatment conditions, qRT-PCR revealed similar trends in PR-B and total PR mRNA expression, with the exception that Dex appeared to diminish slightly PR transcript levels in the presence of 17β-E2, although this did not reach the level of statistical significance (P > .05, Figure 5C). Immunoblot analysis indicated that Dex alone did not affect basal PR protein expression nor did it alter 17β-E2–induced, whereas MPA and Org-2058 both abrogated 17β-E2–induced PR expression (Figure 5D and E). Although PR-A expression at the protein level appeared to be slightly upregulated by Dex in E2-primed cells, this did not reach the level of statistical significance (Figure 5C). In all instances, we observed that MPA and Org-2058, but not Dex, slightly increased PR protein and mRNA expression in the absence of 17β-E2 priming.
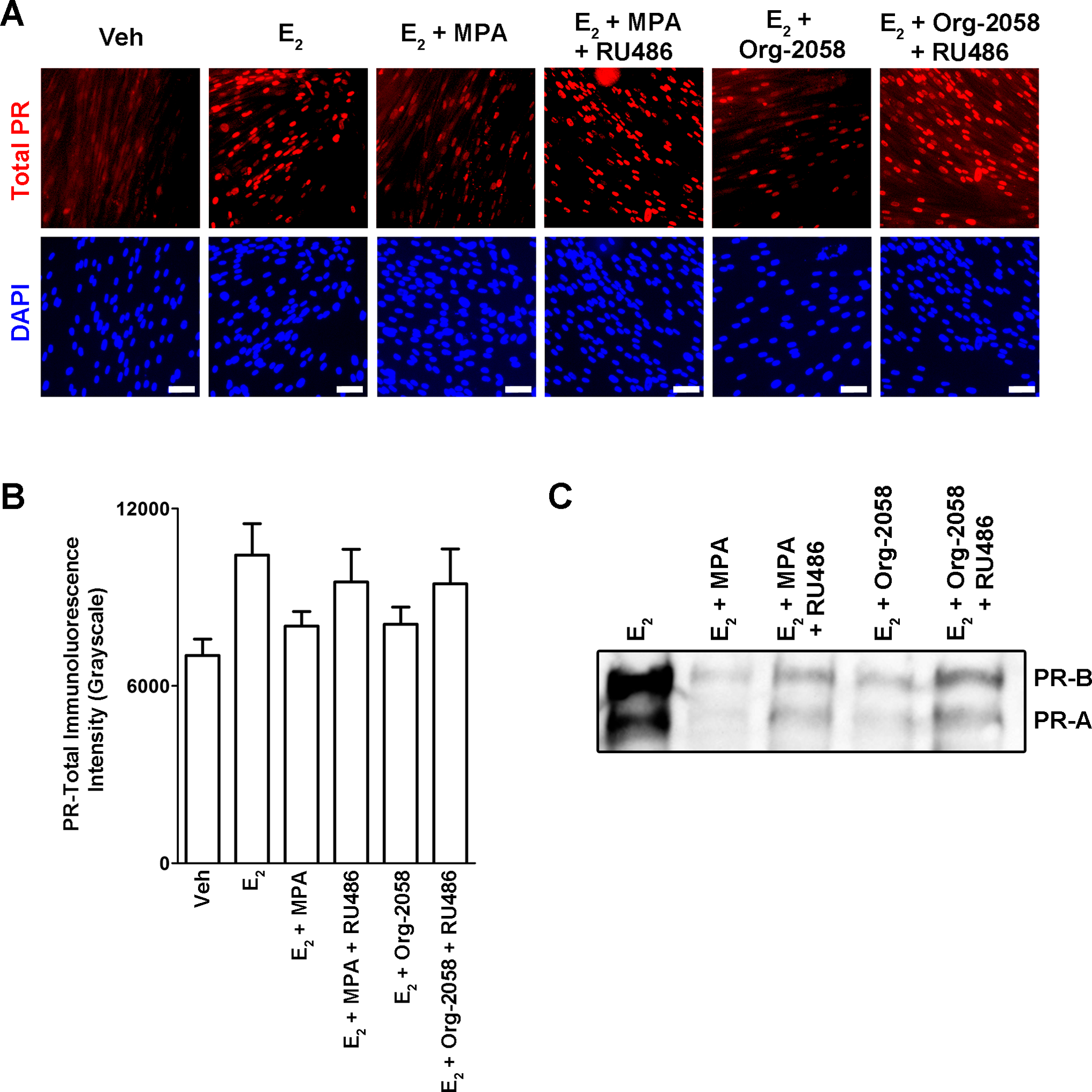
RU-486 attenuates PR agonist-induced PR downregulation in the presence of 17β-E2. A, Cervical fibroblasts were treated for 7 days in the presence of solvent control (Veh, 0.01% ethanol), 10−8 mol/L 17β-E2, 17β-E22 with 10−7 mol/L MPA in the absence or presence of 10−6 mol/L RU-486, or 17β-E2 with 10−7 mol/L Org-2058 in the absence or presence of RU-486. Representative immunofluorescence images are shown for cervical fibroblasts immunolabeled with anti-PR (red) and counterstained with DAPI (blue). Scale bars = 20 μm. B, Bar graph (mean ± SD) summarizing fluorescence intensity measurements for total PR-like immunoreactivity for experiment described in panel A. C, Immunoblot of cervical fibroblast nuclear extracts treated identically as in panel A, probed using an anti-PR antibody
To determine whether downregulation of PR expression by MPA and Org-258 could be reversed by antagonizing binding to the PR, additional experiments were performed using the PR antagonist RU-486. Immunofluorescence and immunoblotting observations revealed that RU-486 partially restored PR expression during coincubations with 17β-E2 and either MPA or Org-2058. Taken together, our results suggest that the downregulating effects of MPA on PR protein expression are selective for the PR and does not involve the GR.
Discussion
The reemergence of P4 as a therapeutic tool to prevent preterm birth has led to increased interest in understanding PR function in reproductive tissues during both pregnancy and parturition. As part of a larger initiative to understand the role of P4 receptor signaling in preterm birth, in the current work, we developed an in vitro model of the cervical stroma, the principal site of mechanical remodeling during preparation for birth. 35 -38 While aspects of functional P4 withdrawal have been described in the human myometrium and decidua, few studies have examined this process in the human uterine cervix. 6 The role of P4 in the maintenance of cervical integrity is supported by studies in both humans and numerous animal models, whereby administration of antiprogestins promoted cervical ripening. 15,38 -43 Randomized controlled clinical trials demonstrated that administration of P4 slowed the rate of cervical shortening in women at risk of delivering premature relative to those who did not receive hormone supplements. 44,45 The mechanisms through which P4 and synthetic progestins maintain cervical integrity, or how cervical ripening may proceed in the presence of high circulating P4 levels are unknown.
In the present work, we found that cervical fibroblasts cultivated in vitro constitutively expressed ER-α but, unlike the corresponding cells in intact premenopausal cervices, did not constitutively express nuclear PRs. Such PR mRNA and protein expression could be restored following incubation with 17β-E2, which is consistent with observations in other estrogen-responsive tissues. 46 -48 When E2-priming was combined with the mixed PR/GR agonist MPA, PR expression was downregulated relative to 17β-E2 treatment alone. This was P4 dependent, inasmuch as this was replicated using the PR-selective agonist Org-2058, but not the GR-selective agonist Dex. Additionally, progestin-induced PR downregulation was abolished by coincubation with the PR antagonist RU-486. These observations are consistent with studies in breast cancer cell lines, where amino acid radiolabeling studies demonstrated that P4 reduced the half-life of its receptor. 49 These effects were mediated by posttranscriptional signaling leading to proteasome-mediated degradation of ligand-occupied PR. 50 It is not clear whether similar mechanisms are active in cervical fibroblasts. Our findings underscore a general theme in nuclear receptor signaling that numerous steroid (including ER, PR, and GR) and nonsteroid receptors (including thyroid hormone receptor, retinoid receptors, and peroxisome proliferator-activated receptor–γ) receptors undergo ligand-mediated proteolysis. 51,52 The seemingly counterintuitive downregulation of receptors by their cognate ligands has generated much speculation as to its biological significance 51,52 ; possibly, coupling nuclear receptor activation with degradation could serve to rapidly “reset” a cell’s transcriptional program in the face of fluctuating hormone levels. In addition, it is possible that P4 receptor downregulation contributes to functional P4 withdrawal in reproductive tissues.
In addition to ligand-induced PR downregulation, we found that in the absence of 17β-E2 priming (with attendant PR upregulation), both MPA and Org-2058 (but not Dex) induced PR expression. These results are in keeping with studies of endometrial stromal cells in vitro, 53,54 in which PR expression was induced by progestins. Our results suggest that, while low, the basal levels of PR expression in vehicle-treated cervical fibroblasts were sufficient to drive PR upregulation. The mechanistic basis for PR induction by progestins has been unclear, as the proximal upstream promoter of the PR gene lacks a canonical PR response element. Tang and colleagues observed that PR may transactivate PR in combination with specificity protein 1 via an Sp1 response element in decidual cells, 54 and it is possible that a similar mechanism occurs in cervical fibroblasts. Collectively, these results suggest that P4 exerts complex and opposing effects on the expression of its cognate receptor, simultaneously licensing and constraining cellular responses to this hormone.
When considered in the context of other investigations, 55 our results have important implications for the study of PR activity in cervical fibroblast cultures, inasmuch as nuclear PR expression cannot be assumed in all settings. As a case in point, in their study of the effects of P4 on lipopolysaccharide (LPS)-induced cytokine secretion, there was no indication that Fukuyama et al used estrogen priming to restore PR expression in their cervical fibroblast models. 56 A comparable study by Kim et al found that in the apparent absence of 17β-E2 priming, 10−6 mol/L P4 (but not lower concentrations) abrogated LPS-induced interleukin-6 expression in fibroblasts derived from pregnant cervical biopsies. 57 Had these authors studied the anti-inflammatory effects following 17β-E2–induced PR expression, it is likely that different effects would have been observed. Another possibility is that these effects could have been mediated through another nuclear receptor such as GR, 58 although the binding of natural P4 to this receptor is relatively weak. 59 It is worth noting that the above studies used P4, rather than the mixed PR/GR agonist, MPA, and as such, it is less likely that anti-inflammatory effects might have been mediated through GR as was recently observed in human uterine smooth muscle cells. 60
That GR and PR exhibit ligand promiscuity is unsurprising in light of molecular phylogenetic evidence. Sequence analyses revealed that the adrenal and sex steroid receptors cluster within a distinct clade in the nuclear receptor superfamily, deriving from a common estrogen-sensitive ancestral receptor. 61 Duplication and divergence of this estrogen-sensitive predecessor yielded a common antecedent for the 3-ketosteroid receptors (ie, PR, AR, GR, and the mineralocorticoid receptor, MR). 61 Recently, Eick and Thornton computationally inferred the coding regions of these ancestral steroid receptors with high confidence, then synthesized cDNAs corresponding to their respective ligand binding domains for functional analyses. 62 These authors demonstrated that the common ancestral receptor was highly sensitive to estrogens (including estradiol, estrone, and estriol), but unresponsive to a panel of corticosteroids, progestogens, androgens, and mineralocorticoids. In contrast, the ancestor of the 3-ketosteroid receptor subclass lacked estrogen responsiveness but was strongly activated by numerous progestins and corticosteroids. The authors noted that while natural selection through gene duplication events and subsequent molecular refinements has increased the selectivity of PR, AR, and GR for their respective naturally occurring ligands (MR remains highly sensitive to endogenous glucocorticoids), 63 this selectivity is imperfect, particularly for synthetic ligands such as MPA and RU-486. For these reasons, caution should be employed when interpreting experimental results following the use of certain synthetic PR agonists and antagonists in the study of PR function.
Results from the current studies indicate that progestin agonists can downregulate PR expression in cervical fibroblasts and may have implications for P4 actions in this tissue. Although the mechanism of functional P4 withdrawal leading to parturition is by no means understood, we postulate that PR receptor modulation may be part of this scheme. Future studies in our laboratory are directed at understanding the role of PR expression levels in the context of cervical maturation. It is premature to speculate on the exact impact of PR downregulation by its agonists, but certainly our data support a role for progestins in downregulating receptor expression at the end of pregnancy and perhaps in the preterm setting.
Footnotes
Acknowledgments
The authors gratefully acknowledge the Ohio State University Wexner Medical Center (OSUWMC) Cooperative Human Tissue Network for assistance with collection of cervical tissues, as well as the technical support of the OSUWMC Pathology Core Facility in assisting with histological sectioning.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Grant Support: This work was supported by the
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.