
Abstract
This study aimed to scrutinize the antifibrotic effects of jujuboside A-loaded exosomes (JuA-Exo) on TGF-β1-induced MRC-5 fibroblasts and explore the pathophysiological basis. Exosomes were isolated from human mesenchymal stem cells and loaded with JuA by ultrasonic dispersion. A TGF-β1-induced MRC-5 fibrosis model was established. Cell viability, migration capacity, levels of inflammatory cytokines, extracellular matrix components, and autophagy-related markers were assessed. To determine autophagy dependence, the autophagy inhibitor 3-methyladenine (3-MA) was employed. JuA-Exo significantly attenuated fibroblast activation triggered by TGF-β1, decreased the secretion of IL-6, IL-1β, and hydroxyproline, and downregulated fibrotic gene expression, including α-SMA, fibronectin, COL1A1, and COL3A1. Mechanistically, JuA-Exo was associated with modulation of the TGF-β/Smad signaling pathway and restoration of autophagy-related markers. Importantly, the defensive effects of JuA-Exo were reversed upon autophagy inhibition by 3-MA. JuA-Exo exerts antifibrotic and anti-inflammatory effects, which are associated with modulation of TGF-β/Smad signaling and autophagy-related processes; however, the mechanistic linkage between these pathways and their causal roles requires further validation. These findings highlight JuA-Exo as a potential experimental candidate, although the current evidence is limited to in vitro observations and does not yet support therapeutic application.
Impact Statement
This study demonstrates that jujuboside A-loaded exosomes exert antifibrotic and anti-inflammatory effects in vitro, providing mechanistic insight and highlighting a potential nanotherapeutic approach that warrants further validation.
Introduction
Pulmonary fibrosis (PF) represents a persistent and worsening interstitial lung disorder, marked by aberrant fibroblast expansion, transition into myofibroblasts, and abnormal accumulation of extracellular matrix (ECM), which collectively contribute to lung structural remodeling and progressive respiratory dysfunction.1,2 Idiopathic pulmonary fibrosis (IPF), accounting for the majority of PF cases, is characterized by a median postdiagnosis survival of just 3–5 years. Epidemiological studies indicate a rising global incidence of IPF, particularly among elderly populations, with rates ranging from 2 to 30 cases per 100,000 individuals annually.3,4 Although antifibrotic agents such as pirfenidone and nintedanib can decelerate disease progression, their clinical benefits remain limited and are frequently accompanied by adverse effects. These shortcomings highlight the pressing demand for innovative and improved treatment approaches that directly target the core mechanisms underlying pulmonary fibrogenesis.
Among the diverse signaling pathways implicated in PF, the fibrogenic factor transforming growth factor-beta (TGF-β)/Smad signaling axis plays a central role in initiating and sustaining fibrotic responses. 5 This pathway promotes fibroblast activation, epithelial-to-mesenchymal transition (EMT), and ECM synthesis. 6 In parallel, emerging evidence highlights the importance of autophagy in fibrotic diseases. Autophagy, a cellular degradation process, is essential for maintaining homeostasis by eliminating damaged organelles and misfolded proteins. Disruption of autophagic flux has been shown to exacerbate fibrogenic processes, whereas its restoration can attenuate fibrosis in the lung and other organs.7,8 In addition, exosomes derived from mesenchymal stem cells (MSCs) are emerging as promising therapeutic tools due to their ability to transport bioactive molecules, combined with high biocompatibility, low immunogenicity, and efficient drug delivery capacity. 9
Jujuboside A (JuA) is a triterpenoid saponin compound extracted from the seeds of Ziziphus jujuba var. spinosa, a traditional Chinese medicinal plant. Previous studies have demonstrated that JuA exerts significant anti-inflammatory and antioxidant effects by modulating NF-κB and Nrf2 signaling pathways, thereby reducing proinflammatory cytokine production and oxidative stress.10,11 Given that chronic inflammation and oxidative stress are critical drivers of pulmonary fibrogenesis, 12 these characteristics suggest a potential rationale for investigating JuA in the context of fibrosis. Moreover, a recent study has revealed that JuA can ameliorate tubulointerstitial fibrosis in diabetic mice by downregulating the YY1/TGF-β1 signaling pathway, a central axis in tissue fibrosis. 13 This finding provides direct experimental evidence for the antifibrotic potential of JuA beyond its established anti-inflammatory effects. Although these findings suggest therapeutic potential for JuA in chronic diseases, its role in fibrotic conditions—particularly PF—remains poorly characterized. Furthermore, clinical application of JuA is constrained by its poor aqueous solubility and low oral bioavailability, challenges that may be overcome through incorporation into nanocarrier systems such as exosomes.
In this study, we aimed to scrutinize the antifibrotic effects of jujuboside A-loaded exosomes (JuA-Exo) in a TGF-β1-induced MRC-5 human lung fibroblast model. We systematically assessed the physicochemical properties of JuA-Exo, including its morphology, particle size, and drug release profile. In addition, we evaluated its cellular uptake, cytotoxicity, and biological activity. Specifically, we examined the effects of JuA-Exo on fibroblast activation, migration, autophagy, and the TGF-β/Smad signaling pathway. To verify whether autophagy is essential for its antifibrotic action, we also employed the autophagy inhibitor 3-methyladenine (3-MA). Collectively, this work provides mechanistic insight into JuA-Exo function, laying the foundation for future in vivo studies to evaluate its translational potential.
Materials and Methods
Cell culture and experimental grouping
Human embryonic lung fibroblasts (MRC-5, CCL-171™) were supplied by the American Type Culture Collection (ATCC, Manassas, VA) and cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Cat#11965092), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin at 37°C in a 5% CO2 incubator. Cells were passaged at 70–80% confluence using 0.25% trypsin-EDTA.
For experimental treatments, cells were divided into five groups: (1) Control, without treatment; (2) TGF-β1 group, treated with 10 ng/mL TGF-β1 for 24 h; (3) TGF-β1 + JuA group, cotreated with 100 μM JuA; (4) TGF-β1 + JuA-Exo group, cotreated with JuA-loaded exosomes; and (5) TGF-β1 + JuA-Exo + 3-MA group, pretreated with 1 mM 3-MA for 1 h before JuA-Exo and TGF-β1 treatment. All interventions were conducted in serum-free medium. JuA-Exo was prepared by ultrasonic loading of JuA into human MSC-derived exosomes, followed by ultracentrifugation to remove free drug. Exosome yield and drug loading efficiency were not quantitatively determined in this study.
Transmission electron microscopy of exosomes
The ultrastructure of MSC-derived Exo and JuA-Exo was visualized using transmission electron microscopy (TEM). Exosome samples were immobilized in 2% paraformaldehyde and deposited onto carbon-coated copper grids. After washing with PBS, the grids were subjected to negative staining using 2% uranyl acetate (Solarbio, Cat#G8170), followed by air-drying. Images were captured using a TEM system (JEOL JEM-2100, Japan) at an acceleration voltage of 100 kV.
Nanoparticle tracking analysis
The morphological dispersion and particle count of Exo and JuA-Exo were assessed using a ZetaView PMX 110 instrument (Particle Metrix, Germany) with nanoparticle tracking analysis (NTA) software. Samples were resuspended in PBS to obtain the target concentration (107–109 particles/mL) prior to analysis. Each sample was measured three times, and the mean diameter was calculated.
Western blot analysis
Exosomes and MRC-5 cells were lysed in RIPA buffer (Beyotime, Cat#P0013B) containing inhibitors of proteases and phosphatases for total protein extraction. A BCA kit was used to measure protein concentration (Thermo Fisher Scientific, Cat#23227). Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes (Millipore, Cat#IPVH00010). After blocking with 5% nonfat milk, membranes were incubated overnight at 4°C with the following primary antibodies: TSG101 (ab125011), CD63 (ab134045), Calnexin (ab22595), TGF-β1 (ab215715), Smad2/3 (ab202445, Abcam), p-Smad2/3 (ab276140), Smad7 (ab216428), LC3B (ab192890), p62 (ab109012), Atg5 (ab108327), and Beclin-1 (ab210498) (all from Abcam). GAPDH (ab8245, Abcam) was included as a loading control. HRP-conjugated secondary antibodies (Jackson ImmunoResearch) were applied, and signal detection was performed using enhanced chemiluminescence (ECL; Thermo Fisher Scientific, Cat#32106). Band intensity was quantified with ImageJ software.
Drug stability assay of JuA
To evaluate drug stability, free JuA and JuA-Exo were incubated in PBS (pH 7.4) at 37°C in the dark. Aliquots were collected at 0, 1, 2, 4, and 8 h, and JuA concentrations were determined by UV spectrophotometry at 279 nm. Relative degradation was calculated by comparing each time point to baseline (0 h), following a previously described method. 14
In vitro drug release study
The liberate profile of JuA from JuA-Exo was monitored using a dialysis bag diffusion method. JuA-Exo samples were placed in dialysis bags (MWCO 10 kDa; Spectrum Labs) and placed in PBS buffer (pH 7.4, 37°C) and gently agitated. At various predefined time points (0, 1, 2, 4, 8, 12, 24, and 48 h), fractions were collected from the external medium and replaced with fresh PBS. The amount of released JuA was quantified by UV spectrophotometry (279 nm) according to a previously established method. 9
Immunofluorescence analysis
To track cellular uptake, JuA-Exo was labeled with the lipophilic dye Dil (Beyotime, Cat#C1036) following the manufacturer’s instructions. Labeled exosomes (0, 10, 30, 50, 100, and 200 μg/mL) were incubated with MRC-5 cells for 6 h. After fixation with 4% paraformaldehyde, DAPI was used to counterstain the nuclei, and cells were imaged using a confocal fluorescence microscope (Zeiss LSM 880). For autophagosome detection, cells were stained with anti-LC3 antibody (ab192890, Abcam) followed by Alexa Fluor® 488-conjugated goat antirabbit IgG secondary antibody (Abcam, Cat#ab150077). Nuclei were again counterstained with DAPI, and LC3 puncta were visualized and quantified using ImageJ.
Cell viability assay
Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8, Dojindo, Cat# CK04). MRC-5 cells were seeded into 96-well plates and treated with JuA-Exo at a range of concentrations (0–200 μg/mL) or assigned to experimental groups (Control, TGF-β1, TGF-β1 + JuA, TGF-β1 + JuA-Exo). Following 24 h of treatment, cells were exposed to CCK-8 for 2 h, and the absorbance at 450 nm was subsequently determined using a microplate spectrophotometer (BioTek Synergy HTX).
Wound healing assay
MRC-5 cells were cultured in 6-well plates and scraped using a sterile 200 μL pipette tip to generate a linear wound. Upon PBS rinse, cells were subjected to the corresponding interventions in serum-free medium. Wound images were taken at 0 and 24 h under an inverted microscope (Olympus CKX53), and closure areas were measured using ImageJ.
Enzyme-linked immunosorbent assay
Levels of hydroxyproline (HYP), interleukin-6 (IL-6), and interleukin-1β (IL-1β) in culture supernatants were measured using commercial enzyme-linked immunosorbent assay (ELISA) kits (HYP: Jiancheng, Cat#A030-2; IL-6 and IL-1β: MultiSciences, Cat#EK206/2 and EK201B/2). Absorbance values at 450 nm were obtained using a plate reader.
Quantitative real-time PCR
To evaluate gene expression changes, total RNA was isolated from MRC-5 cells using TRIzol reagent (Invitrogen, Cat#15596026). The resulting RNA was reverse-transcribed into cDNA with a commercial RT kit (Takara, Cat#RR047A), and transcript levels were quantified by SYBR Green-based real-time PCR on a CFX96 platform. GAPDH served as the internal control, and relative mRNA levels were calculated using the comparative Ct (2−ΔΔCt) method. The primer sequences are listed in Table 1.
Primer Sequences Used for qRT-PCR

Statistical analysis
All experiments were independently repeated three times unless noted otherwise, and results are expressed as the mean ± standard deviation. Statistical analyses were conducted using SPSS software (version 23.0; IBM Corp., Armonk, NY). Differences between two independent groups were evaluated using the unpaired Student’s t-test. For comparisons involving more than two groups, one-way analysis of variance followed by Tukey’s post hoc test was employed. A p value less than 0.05 was considered indicative of statistical significance.
Result
Characterization and stability of JuA-loaded exosomes
TEM imaging showed that both Exo and JuA-Exo exhibited typical bilayer vesicles with round or oval morphology (Fig. 1A). NTA revealed particle sizes ranging from 50 to 160 nm for Exo and 60 to 185 nm for JuA-Exo, indicating a slight increase after JuA loading (Fig. 1B). Western blot confirmed strong expression of exosomal markers TSG101 and CD63, while Calnexin was nearly undetectable, demonstrating high sample purity (Fig. 1C). In PBS at 37°C, free JuA underwent time-dependent degradation, whereas JuA-Exo showed significantly greater stability at 2–8 h (p < 0.01) (Fig. 1D). A dialysis-based release assay indicated an initial burst (∼50%) within 6 h, followed by sustained release up to 90% over 48 h (Fig. 1E).
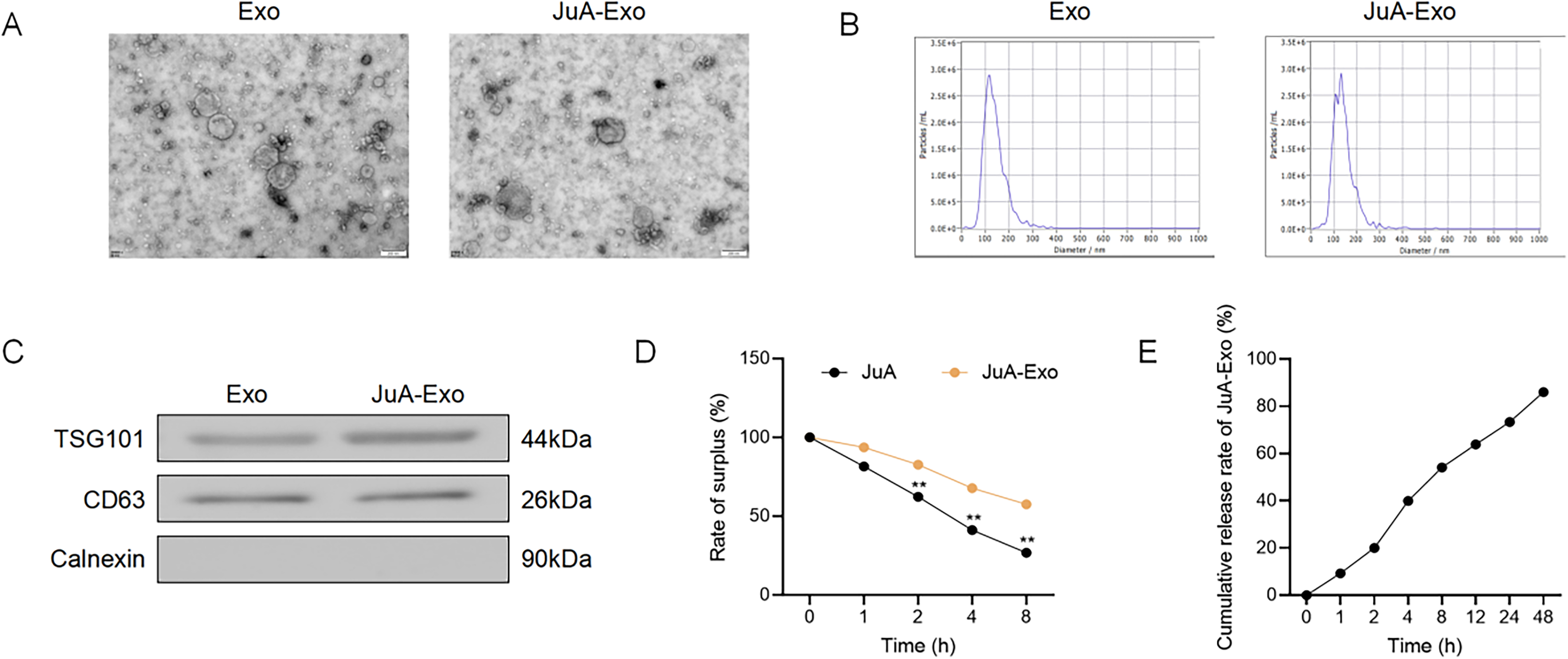
Characterization and release profile of JuA-loaded exosomes.
Collectively, these findings validate JuA-Exo as a well-characterized nanocarrier with improved physicochemical and pharmacokinetic profiles, suitable for subsequent biological evaluation.
JuA-loaded exosomes inhibit TGF-β1-induced cell viability and migration in MRC-5 cells
To assess cellular uptake, MRC-5 cells were incubated with DiI-labeled JuA-Exo at concentrations of 0–200 μg/mL. As depicted in Figure 2A, intracellular red fluorescence increased in a dose-dependent manner, indicating efficient uptake of JuA-Exo, while no signal was detected in the untreated control. The cytotoxicity of JuA-Exo was evaluated using the CCK-8 assay. No significant reduction in cell viability was observed at concentrations up to 100 μg/mL, whereas 200 μg/mL significantly inhibited cell viability (p < 0.01, Fig. 2B), suggesting that JuA-Exo is biocompatible within a therapeutic window. We next examined the effects of JuA and JuA-Exo on TGF-β1-induced fibroblast activation. As expected, TGF-β1 markedly increased MRC-5 cell viability compared with the control (p < 0.01), while both JuA and JuA-Exo treatments significantly reduced this effect. JuA-Exo exerted a more pronounced inhibitory effect than free JuA (Fig. 2C). Wound healing assays further demonstrated that TGF-β1 stimulation enhanced cell migration, which was effectively reversed by both JuA and JuA-Exo. Notably, JuA-Exo treatment led to the greatest reduction in migration rate (Fig. 2D).
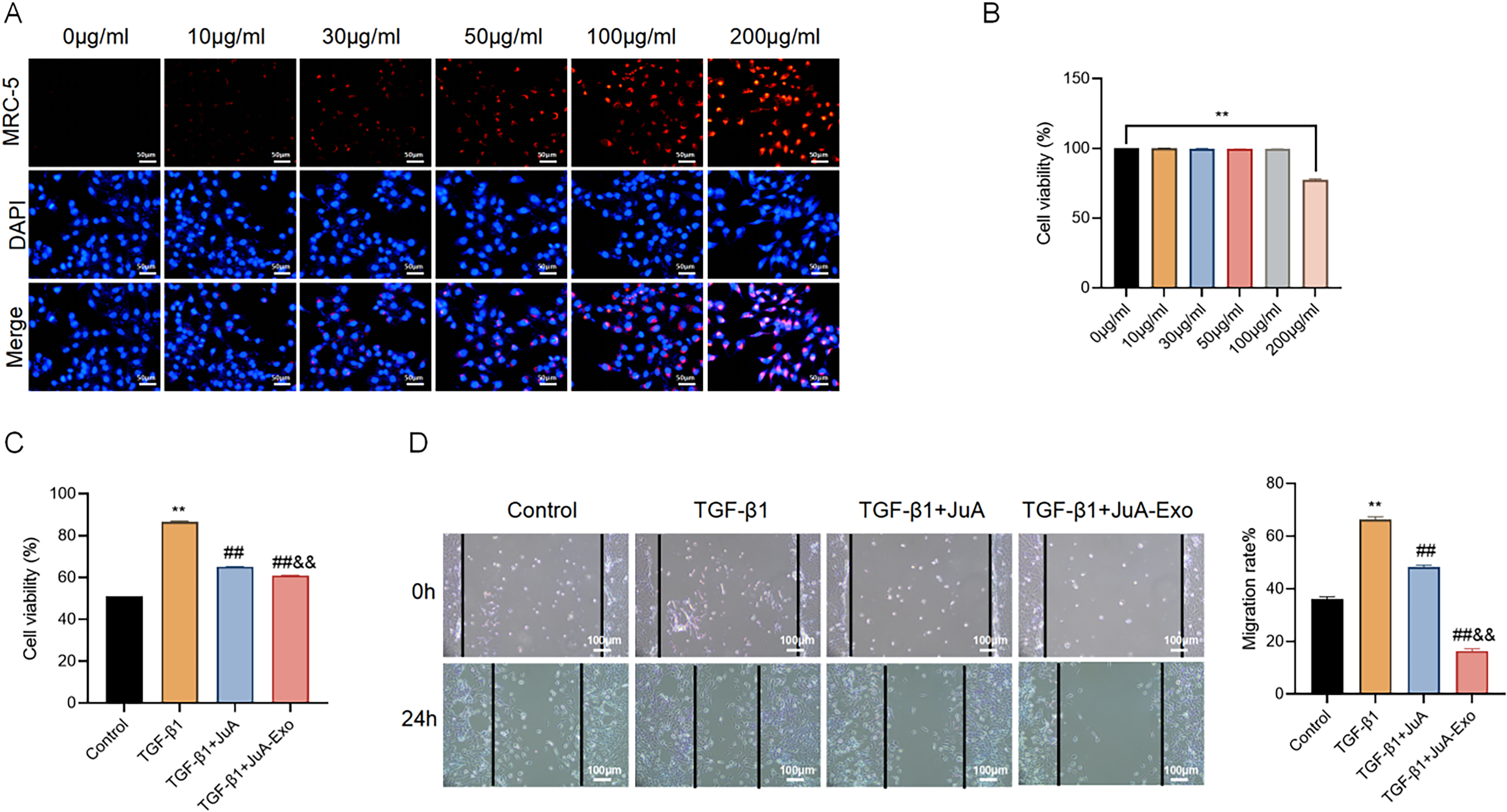
JuA-Exo attenuates TGF-β1-induced viability and migration in MRC-5 cells.
These results indicate that JuA-Exo effectively inhibits TGF-β1-induced fibroblast activation and migration, with enhanced efficacy compared with free JuA.
JuA-Exo attenuates TGF-β1-induced inflammation, fibrosis, and activation of the TGF-β/Smad signaling pathway
To assess anti-inflammatory effects, IL-6 and IL-1β levels were measured by ELISA. TGF-β1 markedly elevated both cytokines, whereas JuA and JuA-Exo significantly suppressed their secretion, with JuA-Exo exhibiting a stronger effect (Fig. 3A, B).
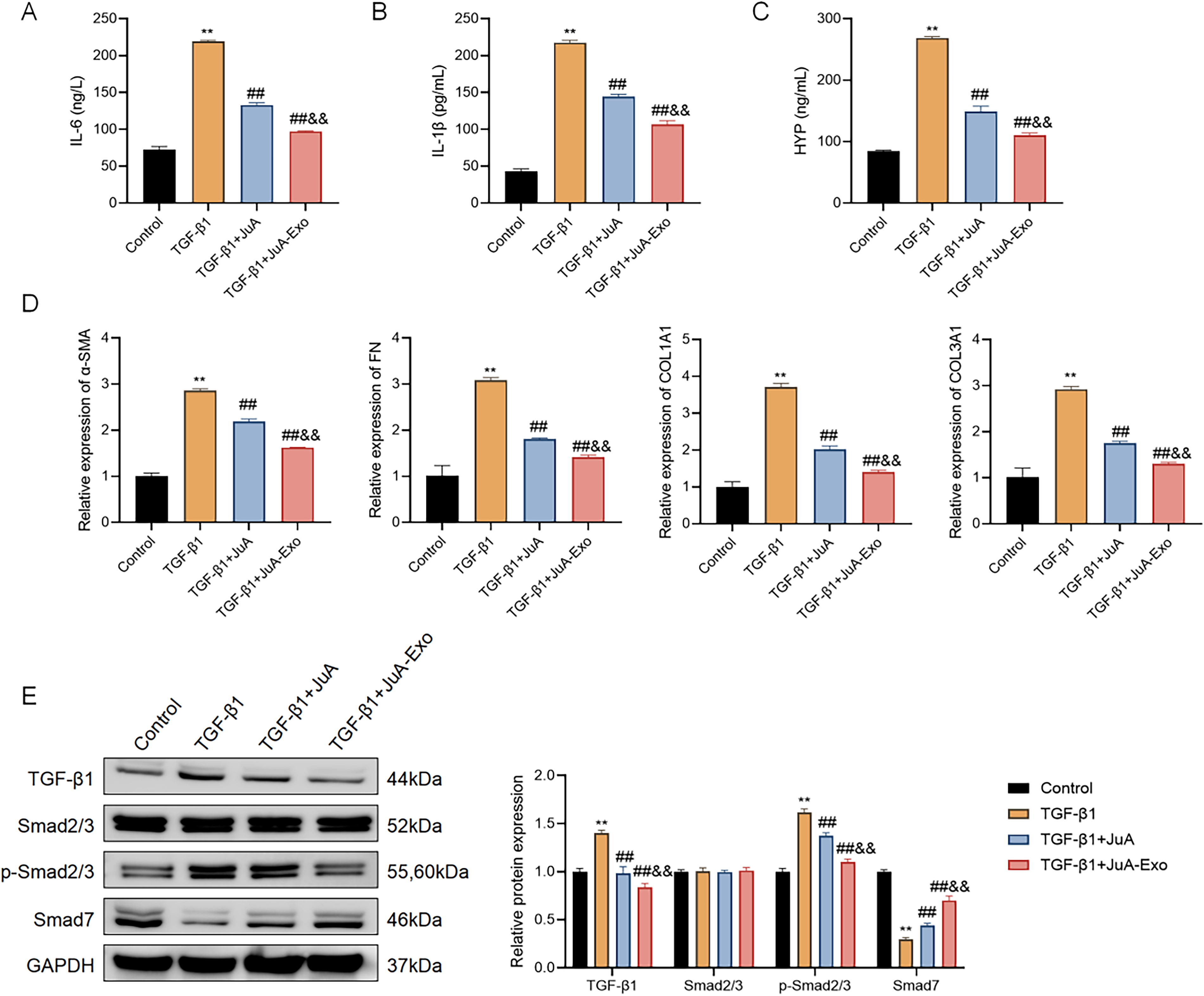
JuA-Exo mitigates TGF-β1-induced inflammatory response, fibrosis, and activation of the TGF-β/Smad signaling pathway in MRC-5 cells. ELISA analysis of IL-6
Similarly, TGF-β1-induced HYP accumulation, indicative of collagen deposition, was reduced by both treatments, with JuA-Exo producing a more pronounced decrease (Fig. 3C). Quantitative real-time PCR (qRT-PCR) analysis showed that fibrotic markers (α-SMA, fibronectin [FN], COL1A1, COL3A1) were markedly increased in response to TGF-β1 and attenuated more effectively by JuA-Exo than JuA (Fig. 3D). To explore the potential mechanism, TGF-β/Smad signaling–related proteins were analyzed by Western blot. TGF-β1 increased the expression of TGF-β1 and p-Smad2/3 and decreased Smad7, without affecting total Smad2/3. JuA-Exo more effectively reversed these changes than JuA (Fig. 3E), suggesting modulation of the TGF-β/Smad signaling pathway at the protein expression level.
These data suggest that JuA-Exo is associated with inhibition of TGF-β1-induced inflammation and fibrosis, potentially involving modulation of the TGF-β/Smad pathway, although direct evidence of Smad transcriptional activity or nuclear translocation was not assessed.
JuA-Exo enhances autophagy in TGF-β1-stimulated MRC-5 cells
To evaluate autophagic activity, LC3 immunofluorescence staining was performed. TGF-β1 treatment markedly reduced LC3 puncta formation, indicating autophagy suppression, whereas both JuA and JuA-Exo restored puncta intensity, with JuA-Exo showing a more pronounced effect (Fig. 4A). Western blot analysis further supported these findings. TGF-β1 decreased the expression of LC3-II, Atg5, and Beclin-1 and increased p62, suggesting suppression of autophagy rather than definitively indicating impaired autophagic flux. JuA and JuA-Exo reversed these changes, with JuA-Exo producing greater upregulation of LC3-II and key autophagy-related proteins (Fig. 4B), suggesting enhanced autophagy activity but not directly confirming increased autophagic flux. Notably, the LC3-II/LC3-I ratio demonstrated a notable increase in the JuA-Exo group when compared with the JuA group, reinforcing its superior ability to enhance autophagosome formation.
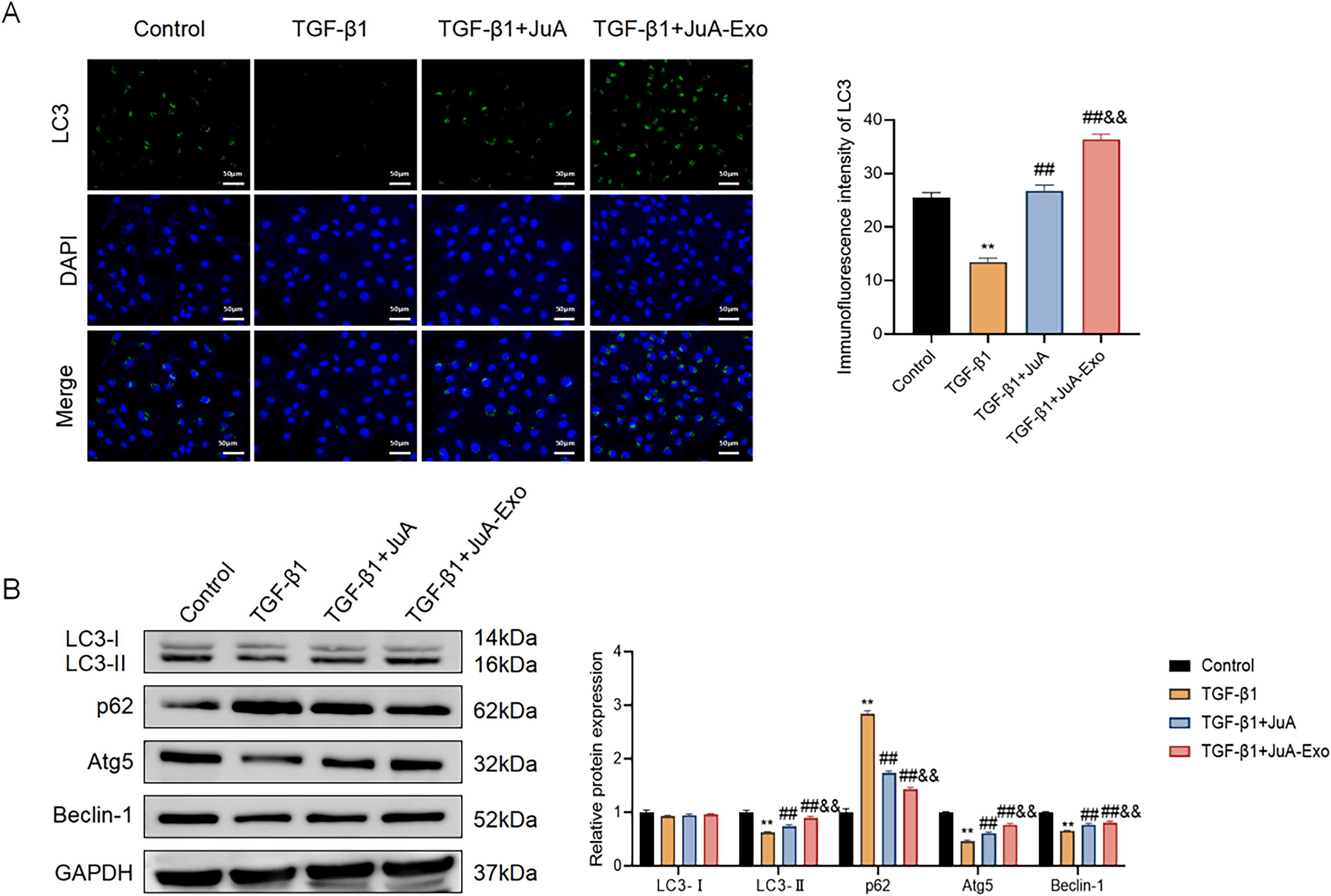
JuA-Exo enhances autophagy in TGF-β1-stimulated MRC-5 cells.
These results demonstrate that JuA-Exo restores autophagic activity more effectively than free JuA in TGF-β1-stimulated MRC-5 cells, although direct assessment of autophagic flux was not performed.
Inhibition of autophagy reverses the anti-inflammatory and antifibrotic effects of JuA-Exo
To determine whether autophagy is essential for the protective effects of JuA-Exo, the chemical inhibitor 3-MA was employed to suppress autophagy in MRC-5 cells under TGF-β1 stimulation. As depicted in Figure 5A, JuA-Exo markedly increased LC3 puncta formation, which was substantially reduced by 3-MA cotreatment, indicating suppression of autophagy. Western blot analysis confirmed these findings (Fig. 5B). JuA-Exo enhanced the expression of LC3-II, Atg5, and Beclin-1 while reducing p62, which is consistent with activation of autophagy but does not directly establish increased autophagic flux in the absence of lysosomal inhibition assays. These effects were partially reversed by 3-MA, as evidenced by downregulation of LC3-II, Atg5, and Beclin-1 and reaccumulation of p62.
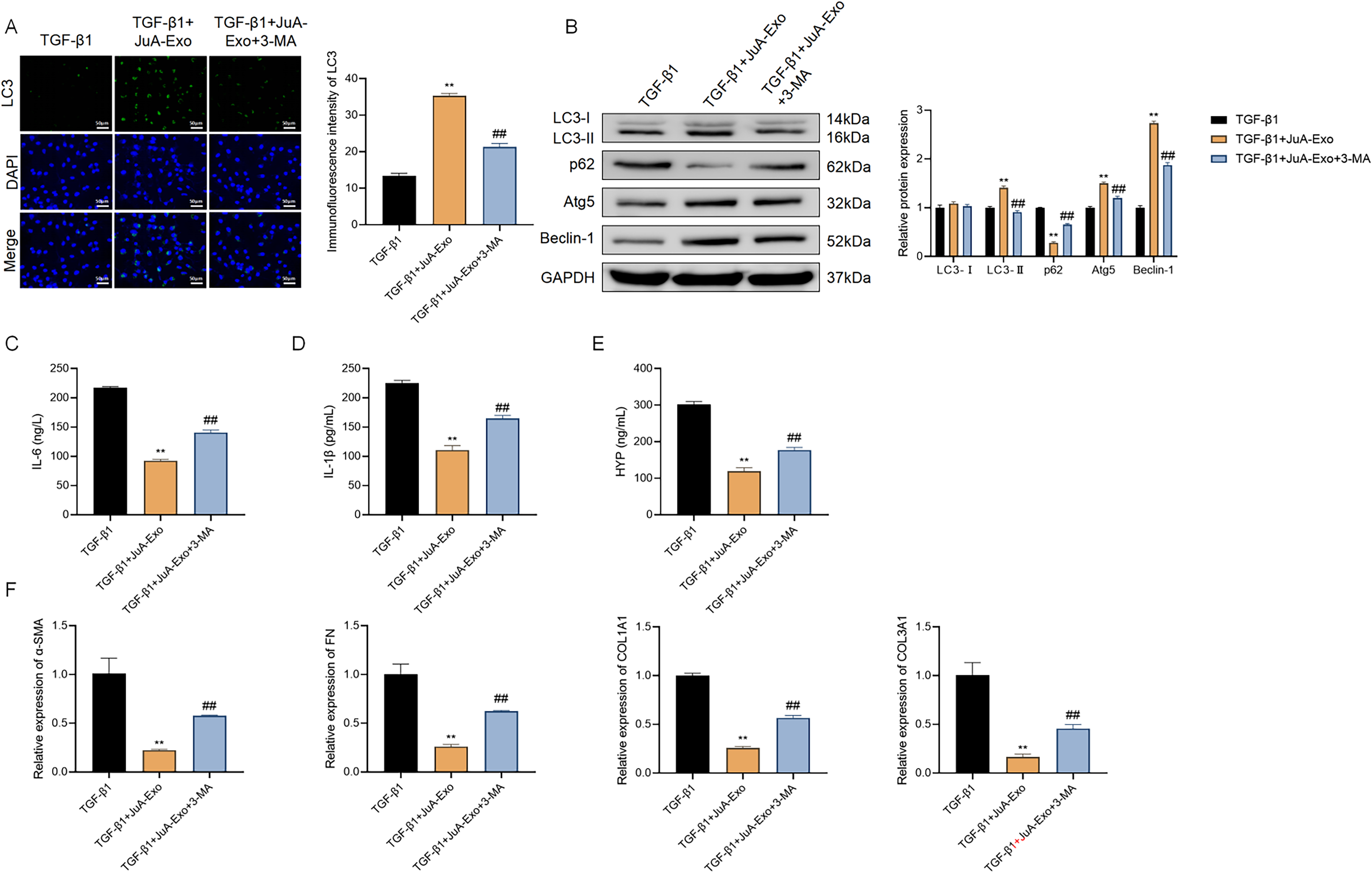
Inhibition of autophagy reverses the protective effects of JuA-Exo in vitro.
Functionally, the anti-inflammatory effects of JuA-Exo were also compromised by autophagy blockade. ELISA revealed that 3-MA significantly restored IL-6, IL-1β, and HYP levels suppressed by JuA-Exo (Fig. 5C–E). Similarly, qRT-PCR analysis showed that JuA-Exo-mediated suppression of fibrotic markers (α-SMA, FN, COL1A1, and COL3A1) was attenuated by 3-MA, indicating a functional dependence on autophagic activity (Fig. 5F).
These results collectively indicate that the anti-inflammatory and antifibrotic effects of JuA-Exo are associated with the activation of autophagy.
Discussion
In this study, we demonstrate that JuA-Exo exerts significant antifibrotic and anti-inflammatory effects in a TGF-β1-induced MRC-5 fibroblast model of PF. Mechanistically, JuA-Exo suppresses the TGF-β/Smad signaling pathway and enhances autophagy-related protein expression. The protective effects of JuA-Exo were partially reversed by the autophagy inhibitor 3-MA, suggesting that autophagy activation contributes to its mechanism of action. However, due to the lack of direct autophagic flux measurement and the nonspecific nature of 3-MA, causal conclusions cannot be drawn. These findings provide in vitro mechanistic insights but do not yet support therapeutic translation.
Consistent with previous reports using exosomes to deliver phytochemicals such as curcumin and quercetin,15,16 JuA-Exo exhibited typical vesicular morphology, nanoscale size distribution, improved drug stability, and sustained release compared with free JuA. These properties address the poor aqueous solubility and low bioavailability of JuA, enabling enhanced cellular uptake and pharmacological activity at lower effective doses. Our data confirm that exosome encapsulation significantly augments the antifibrotic efficacy of JuA, supporting the feasibility of exosome-based nanocarriers for PF therapy.
TGF-β1 stimulation markedly upregulated TGF-β1 and p-Smad2/3 expression while downregulating Smad7, effects that were reversed more effectively by JuA-Exo than by free JuA. These results align with prior studies showing that Smad pathway modulation suppresses fibroblast activation and ECM production.17,18 However, this study did not assess Smad2/3 nuclear translocation or Smad-dependent transcriptional activity (e.g., luciferase reporter assays). Therefore, while JuA-Exo is associated with inhibition of TGF-β/Smad signaling, direct evidence of functional pathway inhibition remains incomplete.
TGF-β1 stimulation reduced LC3-II, Atg5, and Beclin-1 levels and increased p62 accumulation, indicating suppressed autophagy. JuA-Exo reversed these changes, with greater efficacy than free JuA, as reflected by increased LC3 puncta formation and the LC3-II/LC3-I ratio. Nevertheless, these measurements are static and do not distinguish between enhanced autophagosome formation and impaired degradation. Without autophagic flux assays (e.g., using chloroquine or bafilomycin A1), the conclusion that JuA-Exo “enhances autophagy” remains provisional. The data instead support that JuA-Exo restores autophagy-related protein expression under fibrotic stress.
Cotreatment with 3-MA reduced LC3 puncta, downregulated autophagy-related proteins, and attenuated the anti-inflammatory and antifibrotic effects of JuA-Exo. While these observations are consistent with a functional role for autophagy, 3-MA is a nonspecific inhibitor that blocks both class I and class III PI3K, thereby affecting cell survival, proliferation, and TGF-β signaling independently of autophagy.19,20 Thus, the reversal of JuA-Exo’s effects by 3-MA does not constitute definitive proof of autophagy dependence. Future studies should employ more specific approaches, such as genetic knockdown of Atg5 or Beclin-1 or use of lysosomal inhibitors to block autophagic flux, to clarify causality.
Treatment with JuA-Exo significantly reduced secretion of IL-6 and IL-1β, which are key drivers of fibroblast activation and ECM remodeling in fibrotic lung disease.12,21 These effects may be mediated, at least in part, by autophagic clearance of inflammatory mediators, as reported in other injury models.22,23 The dual anti-inflammatory and antifibrotic properties of JuA-Exo further support its potential as a multifunctional experimental candidate.
To our knowledge, this is the first study to construct and characterize JuA-Exo, demonstrating improved aqueous stability, cellular uptake, and sustained release. Furthermore, we provide initial evidence for a dual-target mechanism involving TGF-β/Smad suppression and autophagy restoration, with pharmacological inhibition suggesting functional relevance of autophagy. These findings advance beyond prior studies that typically address either pathway in isolation.
Although in vivo data are lacking, based on the known properties of MSC-derived exosomes, we hypothesize that JuA-Exo may exhibit prolonged circulation half-life, enhanced stability in biological fluids, and preferential accumulation in inflamed or fibrotic lung tissue, potentially due to the enhanced permeability and retention (EPR) effect and homing capabilities of exosomes. Future biodistribution studies using labeled JuA-Exo in bleomycin-induced fibrosis models are required to test these hypotheses.
Several important limitations should be acknowledged. First, this study was conducted entirely in vitro using a single fibroblast cell line (MRC-5), which does not recapitulate the multicellular microenvironment of PF, including alveolar epithelial cells, macrophages, and immune cells. Second, no in vivo validation was performed using established models such as bleomycin-induced PF; therefore, pharmacokinetics, biodistribution, therapeutic efficacy, and immunogenicity of JuA-Exo remain unknown. Third, TGF-β/Smad pathway inhibition was inferred solely from protein expression changes without assessing Smad nuclear translocation or transcriptional activity. Fourth, autophagy was evaluated only through static markers without flux measurement, and the reliance on 3-MA limits mechanistic certainty. Fifth, key parameters of the exosome delivery system, including production yield and drug loading efficiency, were not quantitatively evaluated, which hinders reproducibility and dose–response interpretation. Sixth, the concentration of free JuA used (100 μM) is relatively high, reflecting its poor solubility and cellular uptake, but does not diminish the comparative advantage of JuA-Exo. Finally, other signaling cascades (e.g., PI3K/Akt, MAPK, mTOR) that may intersect with TGF-β/Smad and autophagy were not explored.
To advance these findings, future studies should employ in vivo models to assess the therapeutic efficacy, tissue distribution, and safety of JuA-Exo. Mechanistically, autophagic flux should be directly measured using lysosomal inhibitors, and genetic approaches should be used to establish causality between autophagy and fibrosis suppression. In addition, Smad nuclear translocation and transcriptional activity need to be evaluated, and exosome production parameters should be optimized and reported.
Conclusion
JuA-Exo attenuates TGF-β1-induced inflammation and fibrotic activation in MRC-5 fibroblasts, accompanied by inhibition of TGF-β/Smad signaling and restoration of autophagy-related protein expression. While the data suggest a potential role for autophagy in these protective effects, the lack of autophagic flux validation and the nonspecific nature of 3-MA preclude definitive causal conclusions. These findings position JuA-Exo as a promising experimental candidate for PF, but further in vivo studies and mechanistic validation are required before translational consideration.
Data Availability
The data involved in the present study can be provide under reasonable request.
Authors’ Contributions
J.L. and X.L. designed the research study. J.L. and S.Z. performed the research. Y.S., M.H.G., X.L., and J.L. provided help and advice on experiments. J.L., Y.S., and S.Z. analyzed the data. All authors contributed to editorial changes in the article. All authors read and approved the final article. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Footnotes
Acknowledgments
The authors are grateful to all participants in the present study.
Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
This study did not receive any funding in any form.