
Abstract
Hematopoietic stem and progenitor cells (HSPCs) are precisely organized within specialized bone marrow niches containing hematopoietic and nonhematopoietic cell types. Physiologically accurate human models of this niche environment that include functional HSPCs have been lacking until recently. Using induced pluripotent stem cells (iPSCs) grown in mixed-matrix hydrogels, vascularized three-dimensional human bone marrow organoids (hBMOs) can now be generated that contain mesenchymal, endothelial, and hematopoietic cells, all vital components of the bone marrow niche. While hBMOs have been shown to support engraftment of normal and malignant cells, there are numerous opportunities to use hBMOs for developing a wider experimental toolkit to test gene functions and stress responses. Here, we establish two new applications for the hBMO platform. First, we demonstrate successful engraftment of gene-edited CD34+ cells from healthy donors, enabling direct investigation of gene-specific effects on human hematopoiesis in a defined microenvironment. Gene-edited engrafted HSPCs are maintained in hBMOs for 7 days and undergo multilineage differentiation. Second, we adapted a method to induce stress erythropoiesis—a response to acute anemia in mice and humans—in hBMOs, resulting in robust expansion of immunophenotypically defined hematopoietic progenitors and erythroid populations. These two advances expand the laboratory uses for hBMOs and establish this system as a versatile platform for studying human hematopoiesis and erythropoiesis, stress responses, and gene functions within a physiologically relevant bone marrow microenvironment.
Impact Statement
This project establishes and validates new protocols using human bone marrow organoids (hBMOs) with gene editing technologies, creating a versatile platform to study human hematopoiesis in a physiologically relevant microenvironment. By enabling engraftment of CRISPR/Cas9 gene-edited primary human CD34+ cells and modeling stress erythropoiesis, these strategies address key limitations of animal models and less complex in vitro systems. By modifying the culture conditions, we also extend the hBMO toolkit to study erythropoietic stress, provide a promising new platform for direct interrogation of gene function in the contexts of niche–hematopoietic interactions, and for investigation of paracrine signaling/transcriptional control of regenerative responses in human cells. hBMOs are a discovery tool for human mechanisms underlying anemia, bone marrow failure, and hematological malignancies and provide a translationally relevant system for testing therapeutic strategies.
Introduction
Hematopoietic stem cell (HSC) self-renewal and differentiation into mature blood cells is tightly regulated and occurs within specialized niches in the bone marrow microenvironment (BMM). Single-cell transcriptomics studies combined with spatial imaging, immunofluorescence, conditional knockout systems, and genetic marking analyses in mice have revealed the functional complexity of the niche with hematopoietic cells organized among nonhematopoietic cells.1–3 The HSC niche is very difficult to model in vitro due to the architectural complexity and heterologous cell–cell interactions. Once removed from the BMM, the molecular and functional attributes of HSCs change almost immediately, with substantial loss of activity after 24 h.4,5 The BMM includes mesenchymal stromal cells (MSCs), vascular and neural cells, and hematopoietic progenitors mixed with mature hematopoietic cells. 6 While genetic or functional disruption of nonhematopoietic niche cells profoundly impairs hematopoiesis and promotes hematological diseases,7–9 liquid culture experimental systems fail to recapitulate these interactions in human, three-dimensional (3D) contexts. Niche-dependent release of cytokines and growth factors, including stem cell factor (SCF), thrombopoietin (TPO), and TGF-β promotes HSPC quiescence, proliferation, and differentiation.10–13 How these signals are integrated spatially and the dynamics of change in response to disease or acute stress conditions, remain poorly understood, highlighting a need for the development of organoid platforms to study development and disease.
While mouse models have been widely used to study hematopoiesis due to broad conservation of hematopoietic programs compared to humans, some species-specific differences have limited their ability to capture hematopoietic physiology and disease. Importantly, mouse bone marrow contains different numbers of adipocytes, megakaryocytes, and progenitor blood cell types, suggesting distinctions between the two species.14,15 Hematopoietic disease processes also differ. Mouse models have proved frustrating for faithful recapitulation of the disorganized marrow, including in myelodysplastic syndromes (MDS) and leukemia. 16 Humanized mouse strains have been developed that attempt to circumvent the niche deficiencies in mice. However, these mice models involve complex genetics, lack certain human cell types, and can develop graft exhaustion. 17 Moreover, many of the genetic models that cause MDS in humans do not produce the full spectrum of disease phenotypes seen in patients. 18 In anemia, the hematopoietic system shifts to a process called stress erythropoiesis to produce enough erythroid cells.19–21 Both mice and humans can exhibit extramedullary hematopoiesis (primarily in the spleen) that accelerates erythropoiesis and anemia recovery, although there are species distinctions.22–25 A wide range of stress-specific mechanisms are involved in modulating control of erythropoiesis in the context of anemia.19,26–30 Since humanized mouse models lack representative populations of human erythrocytes, studying human hematopoiesis and anemia responses is done ex vivo. Other methods such as coculturing of hematopoietic cells and MSCs, 3D coculture, and ossicle xenotransplantation systems provide some improvements but still form an incomplete picture of the human BMM.31,32 While many methods have been developed for the study of human hematopoiesis, none of them completely recapitulate the unique BMM.
Recently, 3D human bone marrow organoids (hBMOs) from iPSCs have been generated, which accurately mimic major components of the BMM.33,34 Importantly, this method can reproduce the architecture of the BMM, including its specialized sinusoidal vessels. Single-cell sequencing and immunofluorescence have shown reproducibility of cells of the hematopoietic lineages, MSCs, and endothelial cells by the iPSCs that can be found in specific subsections of the organoid, similar to what is observed in the BMM.33,34 Engraftment of both healthy and malignant hematopoietic cells into the organoids has been successful and allowed for the study of the changes when the BMM is perturbed.33–36 Treatment of organoids with TGF-β induces myelofibrosis. 33 The development of 3D hBMO models provides a plethora of new experimental opportunities for accurate modeling of normal and abnormal hematopoiesis. 37 To date, these models provide the closest representation of human hematopoiesis. The potential applications for hBMOs are broad and remain largely unexplored.
Here, we present protocols to evaluate genetic requirements for human hematopoiesis using gene-edited CD34+ HSPCs engrafted into hBMOs. Gene-edited cells engraft, form, and maintain immunophenotypically defined HSCs along with a range of hematopoietic progenitor cell types and, with the use of labeling dyes, can be distinguished from nonengrafted HSCs. We also present a method to induce “stress erythropoiesis” in the hBMOs. Previous work has successfully created stress-burst-forming unit-erythroid colonies using Bone Morphogenetic Protein-4 (BMP4) and low O2 conditions.20,21 While treatment of hBMOs with low O2 or BMP4 alone altered HSPC and increased the frequency of erythroid populations, a combined treatment increases the mature erythroid pool in hBMOs. hBMO approaches present significant improvements over liquid culture systems to test HSPC phenotypes resulting from genetic loss-of-function or environmental perturbation (e.g., modeling anemia).
Methods
Ribonucleoprotein nucleofection
Gene-edited CD34+ HSPCs were purchased from the HSPC Gene Editing core at the Fred Hutchinson Cancer Research Center. Cells were thawed 2–4 h before nucleofection. Gene editing was conducted according to previously defined protocols for optimized nucleofection of single guide RNA (sgRNA):Cas9 ribonucleoprotein (RNP) complexes. 38 Briefly, two lyophilized single guide RNA oligos per gene (Synthego) were dissolved in Tris-EDTA (pH 8.0) and mixed at a concentration of 100 pmol/µL (50 pmol/µL per guide). RNP complexes were assembled by mixing 1 µL of the sgRNAs with 2 µL of sNLS-SpCas9-sNLS nuclease (25 pmol/µL; Aldevron) and 17 µL of Complete P3 solution (P3 4D-Nucleofector Kit) to a total volume of 20 µL. 2–4 ×105 CD34+ cells (resuspended in P3 + RNP complexes) were nucleofected using the DS-150 program on the 4D Nucleofector (Lonza). Freshly nucleofected cells were transferred to CD34 expansion media (StemSpan SFEM II (Stemcell Tech., 09655) with 100 ng/mL TPO, SCF, interleukin-6 [IL-6], fms related receptor tyrosine kinase 3 [FLT-3]) for 4 h and then cryopreserved in CryoStor CS10 (Sigma).
CRISPR editing analysis
Quantitation of Adeno-Associated Virus Integration Site 1 (AAVS1)-CTRL editing efficiencies was determined using the Interference of CRISPR Editing (ICE) PCR assay, while SAMD1 knockout was partially confirmed by SYBRgreen qPCR. Briefly, genomic DNA (MicroElute Genomic DNA Kit, Omega Bio-Tek) was isolated from cells 48 h postnucleofection. PCR amplification of the region surrounding the AAVS1 cut sites was performed using 50 ng DNA, Phusion High-Fidelity DNA Polymerase with GC Buffer, AAVS-ICE-F/R primers, and 1M Betaine. Size-verified PCR products were column purified (Monarch PCR and DNA Clean-up, New England Biolabs) and submitted for Sanger sequencing with the AAVS-ICE-F primer (Genewiz). Trace files were used to quantify editing using the EditCo tool (https://ice.editco.bio). Due to issues with generating high-quality Sanger sequencing data at the SAMD1 edit site, qPCR assays were used to partially quantify editing efficiencies based on a standard curve generated for absolute quantification. One assay quantified the deletion frequency of the region between the two sgRNAs (primers SAMD1-F1/R1), while a second partially quantified sgSAMD1-166 editing as well as deletion between the sgRNAs (Supplementary Fig. S1A-C; primers SAMD1-F/R).
Generation of human BMOs
Human bone marrow organoids were differentiated from human iPSCs (CVCL_RM92) as previously described.33,34 Briefly, the generation of organoids is performed in five phases. Phase 0 iPSCs are detached and aggregated in StemFlex and Revitacell for 1 day. Phase I is 3 days of mesodermal commitment in APEL2 medium containing 50 ng/mL of BMP4, Fibroblast Growth Factor-2 (FGF2), and Vascular Endothelial Growth Factor A (VEGFA) and 3 μM of the CHIR99021 GSK3 inhibitor performed in 2% O2, 5% CO2, 93% N2. Vascular and hematopoietic commitment is induced for 2 days in Phase II in APEL2 medium containing 50 ng/mL of BMP4, FGF2, and VEGFA and 25 ng/mL of hSCF and FLT-3. At phase III, iPSC aggregates were embedded in mixed collagen hydrogels and cultured in APEL2 medium containing 5 U/mL of heparin, 5% KO serum (ThermoFisher Scientific), and 100 ng/mL of VEGFA, 50 ng/mL of Vascular Endothelial Growth Factor C (VEGFC), FGF2, TPO, erythropoieting (EPO), hSCF, BMP4, and Granulocyte-Colony Stimulating Factor (G-CSF), and 20 ng/mL of IL-3 and IL-6 for 5 days. After 2 days, 1 mL of medium was added to each well containing half the concentration of the cytokines and growth factors. Day 10 hBMOs were distributed into individual wells of U-bottomed 96-well plates and cultured for an additional 2 days in Phase IV medium before engraftment or stress experiments.
Engraftment of gene-edited CD34+ cells
Cryopreserved, nucleofected CD34+ HSPCs were thawed one day prior to engraftment and cultured at 1 × 105 cells/mL in StemSpan serum-free expansion media containing recombinant human FLT-3, SCF, IL-3, and IL-6 (Stemcell Technologies). Alternatively, CD34+ cells can be freshly nucleofected one day prior to engraftment. To label engrafted cells, nucleofected CD34+ HSPCs were washed and incubated with 5 μm of carboxyfluorescein succinimidyl ester (CFSE; Biolegend). Cells were resuspended in Phase IV medium composed of StemPro-34 (Gibco) containing 5 U/mL of heparin, 5% KO serum (Thermo Fisher Scientific), 25 ng/mL of VEGFA, VEGFC, FGF2, hSCF, and FLT-3, and 10 ng/mL of TPO, EPO, IL3, and IL6. 5000 CD34+ cells in 100 μL of Phase IV medium were added to organoid cultures for engraftment. Engrafted organoids were cultured for 7 days. Every 48–72 h, 100 μL of Phase IV media was removed and replaced with fresh Phase IV media.
Induction of “stress” erythropoiesis
Day 12 hBMOs were grown in 100 μL of Phase IV media containing 15 ng/mL of hBMP4 (R&D Systems). Organoids were incubated in either normoxic (20% O2) or hypoxic (2% O2) conditions for 6 days with media changes every 48–72 h.
Dissociation of organoids and flow cytometry
To dissociate organoids into a single-cell suspension, 5–10 organoids per replicate were collected in a 15 mL conical tube using a P1000 trimmed tip. To remove nonengrafted cells, 5 mL of PBS was added and incubated at room temperature for 4 min, then aspirated. Organoids were washed in 10 mL of PBS at 400 g for 5 min. PBS was aspirated, and organoids were resuspended in 500 μL of collagenase (10 mg/mL) in Hank’s Balanced Salt Solution (Corning). Organoids were incubated at 37°C for 6 min and then mixed with a P1000 followed by another 6-min incubation at 37°C. After mixing with a P200, the remaining visible organoids were incubated in 3-min increments at 37°C, followed by mixing with a P1000 until completely dissociated by eye. Immediately after dissociation, 3 mL of 10% FBS in PBS was added to stop enzyme activity. Cells were washed at 200 g for 5 min and resuspended in PBS.
Engrafted and organoid cells were stained with preconjugated antibodies to detect the surface proteins CD34 (clone 561), CD38 (clone HIT2), CD45RA (clone HI100), CD49f (clone GoH3), CD90 (clone 5E10), CD71 (clone CY1G4), CD235a (clone HI264), CD45 (clone 2D1), CD11b (clone M1/70), fixable Live/Dead Blue stain (Thermo Fisher Scientific), and a biotin-conjugated lineage negative cocktail: CD3 (clone OKT3), CD19 (clone HIB19), CD14 (clone M5E2), CD56 (clone HCD56), and CD20 (clone 2H7). Data were collected on the LSR Fortessa and analyzed in FlowJo v10.10.0.
Statistical analysis
Statistics were performed as stated in each figure and were performed in GraphPad Prism v10.4.2. Quantitative data are expressed as +/− standard error of the mean. Significance is defined as *p < 0.05, **p < 0.01, and ***p < 0.001.
Experiment
Efficient engraftment of gene-edited CD34+ cells into human bone marrow organoids
We generated organoids using a 10-day protocol outlined in Figure 1A, according to Khan et al. 33 Between days 10 and 12, we developed new protocols to evaluate engraftment of gene-edited cells and the role of low oxygen/BMP-4 treatment on organoid erythropoiesis (Fig. 1B). On day 12, organoids developed characteristic features, including vessel formation and cells with erythroid morphology within vessels (Fig. 1C). To establish hBMOs as a platform for functional genetic interrogation of human hematopoiesis, we first evaluated whether CRISPR/Cas9-edited CD34+ HSPCs could be efficiently engrafted and maintained within the organoid microenvironment. As proof of principle, CD34+ cells from healthy human donors were edited by RNP nucleofection using two single-guide RNAs targeting the Sterile Alpha Motif Domain-1 (SAMD1) gene, a transcription factor that plays a role in hematopoiesis, erythropoiesis, and megakaryopoiesis, 39 or using guides targeting the AAVS1 locus as a control. AAVS1-CTRL editing efficiencies were quantified by ICE analysis of Sanger sequencing (100% indel frequency; Supplementary Fig. S1). Due to technical issues with Sanger sequencing data covering the SAMD1 edited region, we developed a new method to test editing efficiencies using absolute quantitation of SYBRgreen qPCR data (Supplementary Fig. S1). The deletion frequency for the two sgRNAs targeting SAMD1 were 44.9% and 8.8%, respectively. To confirm loss of SAMD1 protein, edited cells were expanded in StemSpan liquid culture containing cytokines (see methods) for 6 days (Fig. 2A).
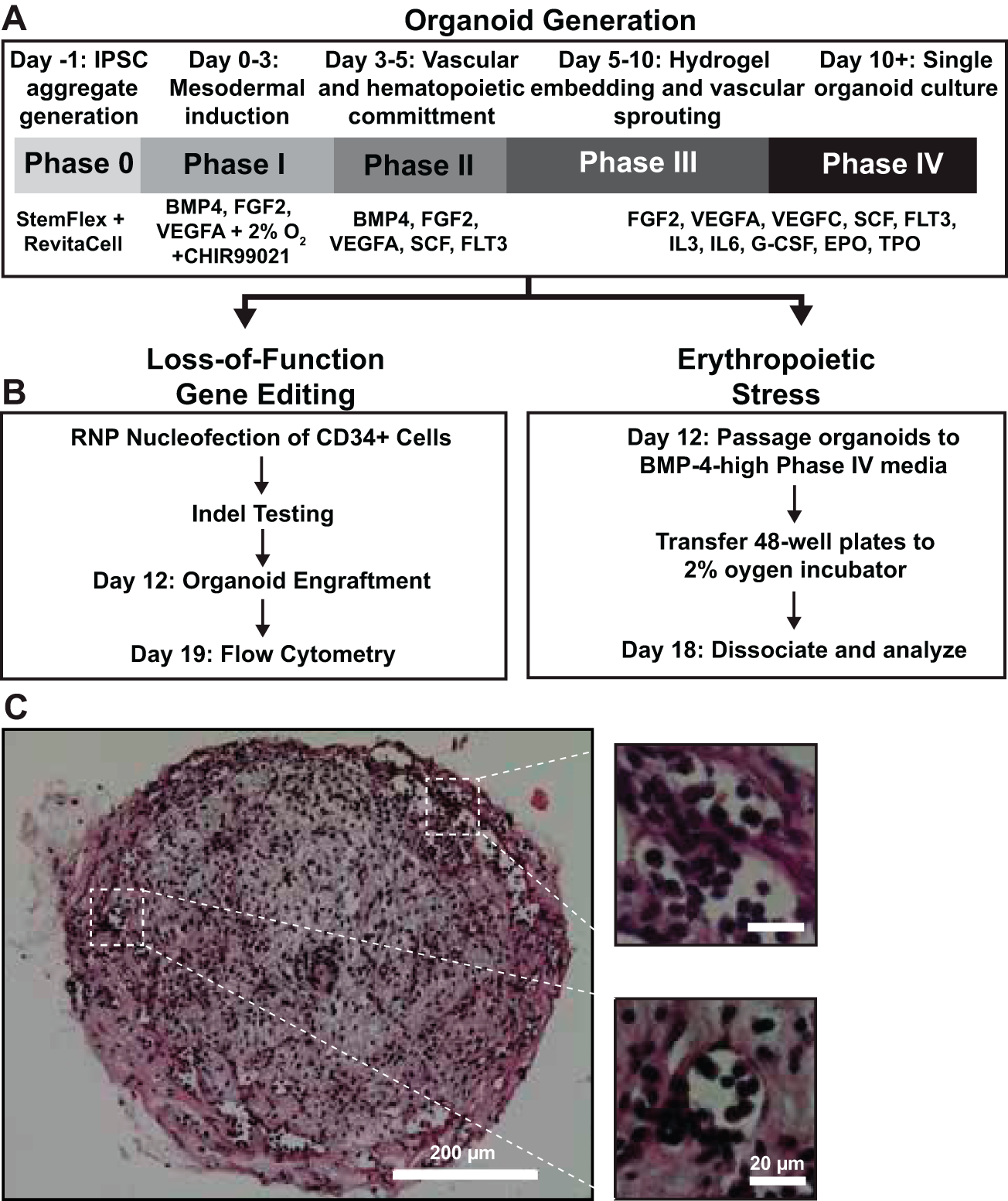
Generation of vascularized organoids for gene loss-of-function experiments and erythropoietic stress.
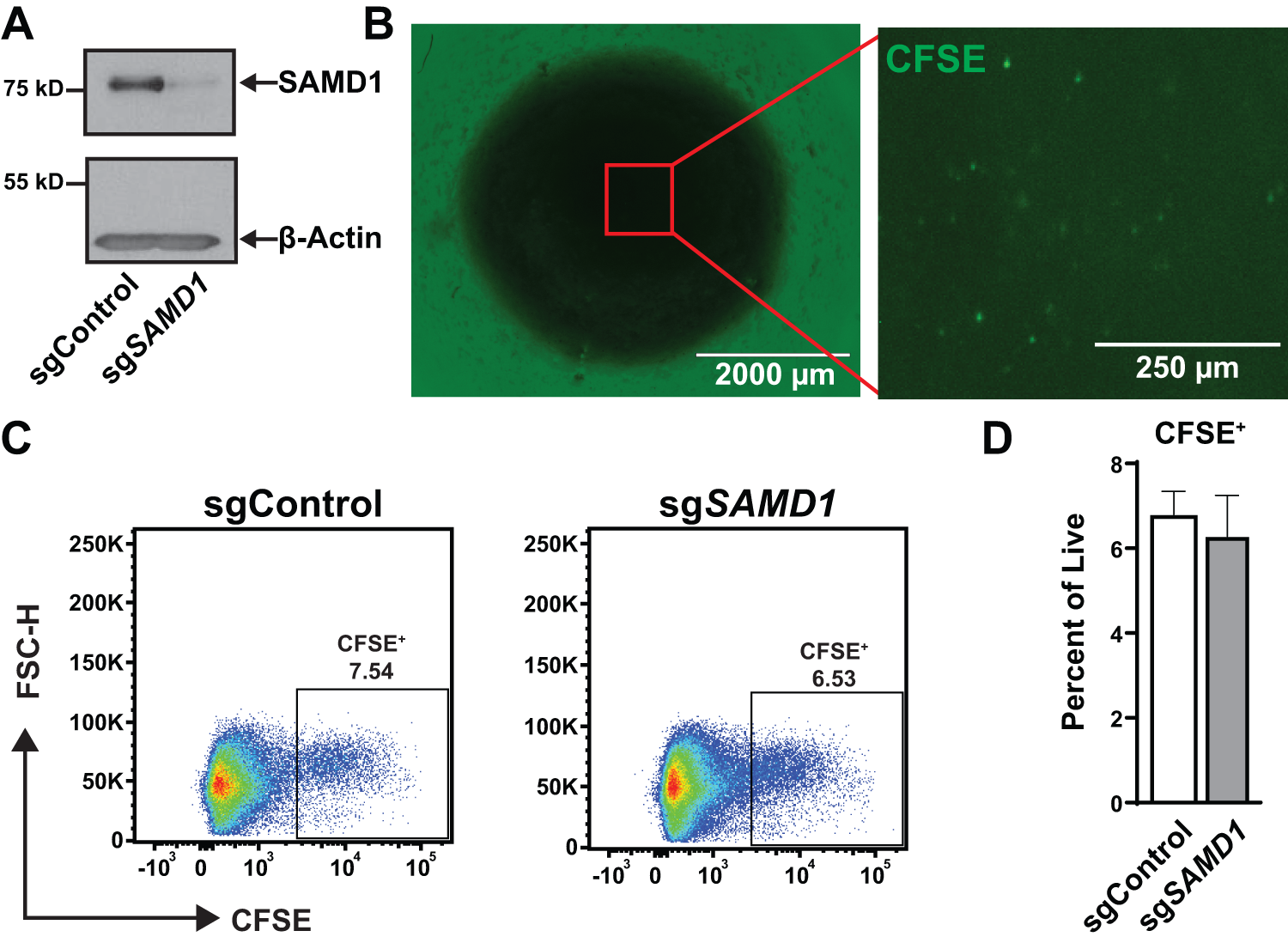
Gene-edited CD34+ HSPCs engraft into hBMOs.
Control and gene-edited (sgSAMD1) CD34+ HSPCs were labeled with CFSE and engrafted into pre-established hBMOs 24 hours after thawing. After 7 days, fluorescence imaging confirmed the presence of CFSE+ cells in the organoid (Fig. 2B). The CFSE+ cells appeared to be spread evenly throughout the organoids and not clustered at one engraftment location. To examine the percentage of engrafted cells in the organoid, hBMOs were dissociated for flow cytometry. For this, 6.5% of the organoid was engrafted cells, and no difference was observed between the cells nucleofected with nontargeting and gene-targeting guides (Fig. 2C and D). These results demonstrate that hBMOs support the engraftment of gene-edited human HSPCs.
Gene-edited CD34+ cells are maintained and undergo multilineage differentiation in hBMOs
To determine whether gene-edited CD34 cells’ retain immunophenotypic HSPC properties in hBMOs, organoids were dissociated after 7 days using collagenase II and manual pipetting for immunophenotypic analysis. Flow cytometric profiling revealed that 10.6% of the engrafted CFSE+ cells were CD34+Lin−, indicating that engrafted, edited immature progenitor populations remained in organoids. The CFSE+CD34+ Lin− population consisted predominantly of common myeloid progenitors (CMPs; CD38+ CD45RA−), with smaller fractions of megakaryocyte/erythroid progenitors (MEPs; CD38low CD45RA−), granulocyte/monocyte progenitors (GMPs; CD38+ CD45RA+), and a detectable populations of HSCs (CD38− CD45RA−, CD90+, and CD49f+), and multipotent progenitors (MPPs; CD38− CD45RA− and CD90− CD49f−; Fig. 3). 40 Common lymphoid progenitors (CLPs) were not observed in the engrafted CD34 population. The lack of CLPs is expected as hBMOs themselves do not support lymphoid differentiation. 33
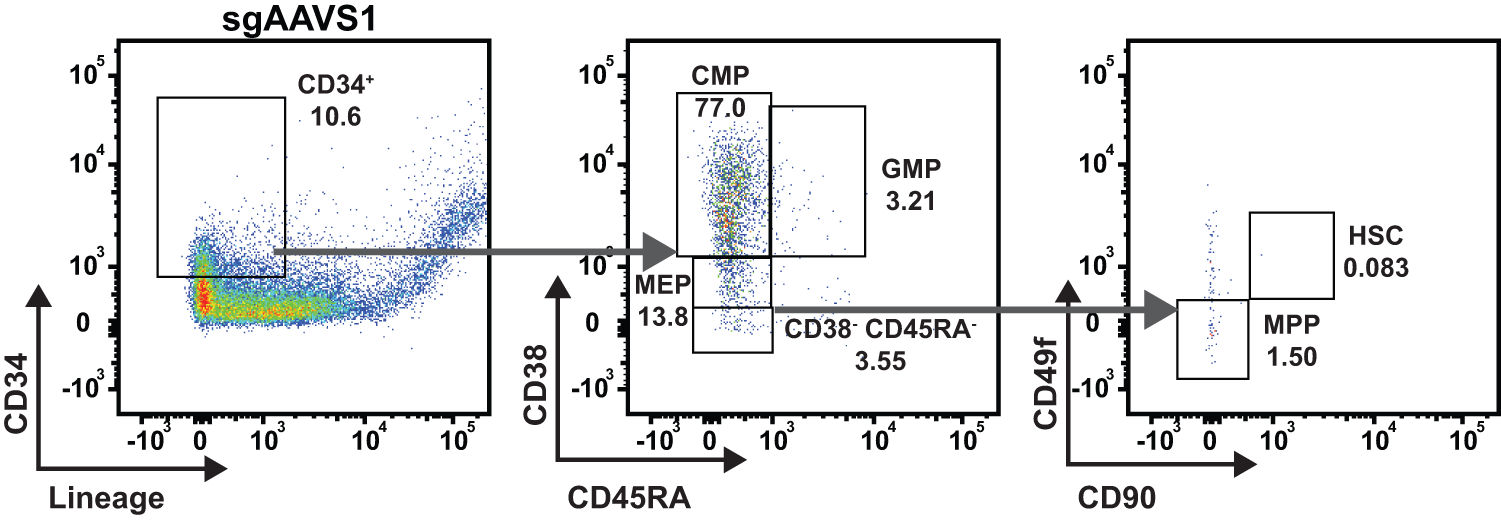
hBMOs support engrafted CD34+ immunophenotypically defined progenitor populations. Representative gating strategy of HSPC cell types in control CD34+ cells nucleofected with sgRNA targeting the AAVS1 locus at day 6: hematopoietic stem/progenitor cells (CD34+ Lin−), CMPs (CD38+ CD45RA−), MEPs (CD38low CD45RA−), and GMPs (CD38+ CD45RA+). CD34+ Lin− shown as percent of CFSE+. CMP, GMP, MEP, CD38− CD45RA−, HSC, and MPP are shown as a percentage of CD34+ Lin−. CMP, common myeloid progenitor; HSC, hematopoietic stem cell; GMP, granulocyte/monocyte progenitors; MEP, megakaryocyte/erythroid progenitors; MMP, multipotent progenitor.
Next, we evaluated whether edited CD34 cells are capable of multilineage differentiation. Analysis of CFSE+ CD45+ cells revealed myeloid differentiation, based on the acquisition of a CD34−CD11b+ population (Fig. 4A). CD71, also known as transferrin receptor, and CD235a can be used to evaluate stages of erythroid maturation. We observed that engrafted CFSE+ cells differentiated to early erythroid progenitors (CD71+ CD235a−) and late erythroid progenitors/mature erythrocytes (CD235a+), making up approximately 20.5% and 27% of CFSE+ cells, respectively. A representative flow panel from three replicates of pooled organoids is shown in Figure 4B. These data demonstrate that hBMOs maintain the HSPC populations of engrafted CD34+ cells after 7 days in culture and also support multilineage differentiation.
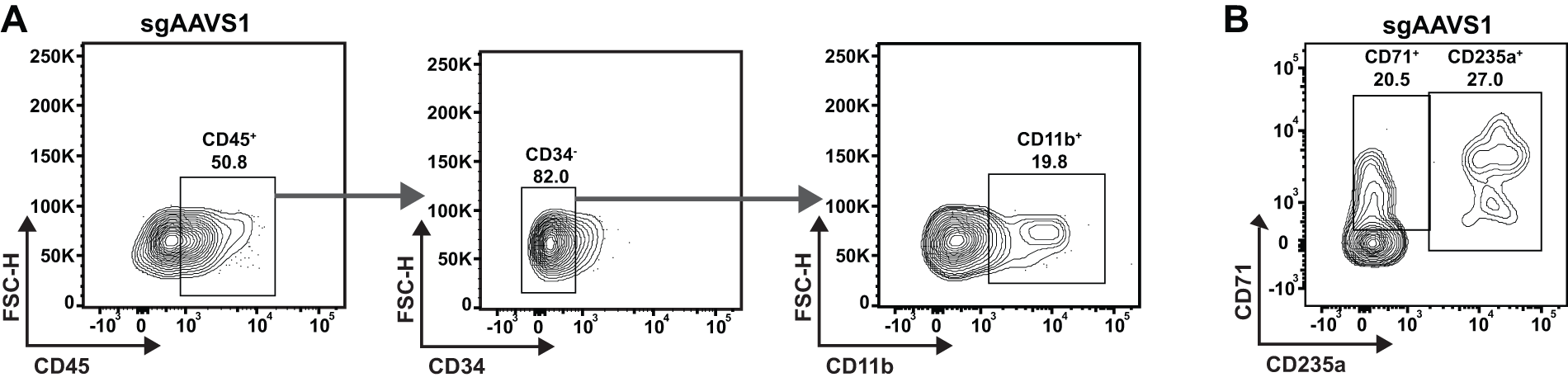
hBMO-engrafted CD34+ HSPCs differentiate to the myelomonocytic and erythroid lineages.
Low O2 and the addition of BMP4 alters hematopoiesis in hBMOs
Stress erythropoiesis occurs during acute anemia in mice and humans to replenish lost erythrocytes, and foundational work showed that BMP4 and O2 levels can mimic this process in liquid cultures ex vivo.20,21 In this light, we tested whether hematopoietic and erythroid progenitor cells in hBMOs can respond to similar environmental signals to increase erythropoietic output. We shifted day 12 organoids to a culture media containing an additional 15 ng/mL BMP4 (or PBS control) and/or low oxygen for 6 days. Pooled organoids were dissociated for comparative flow cytometry analysis. The percentage of Lin−CD34+ cells in hBMOs increased from 4.8% in normoxia to 12.5% in hypoxia (N = 3; Fig. 5A). Hypoxia increased CD34+ cells compared to normoxia in both control media and BMP-4-containing media (Fig. 5B). Next, we analyzed MPP, HSC, CMP, and MEP populations (Fig. 5C). No significant changes were observed in the percentages of progenitor populations within the CD34+ fractions (Fig. 5D). We tested whether the erythroid populations changed in response to hypoxia and/or BMP-4 (Fig. 6A). In liquid cultures, hypoxia and BMP-4 act combinatorially to induce stress erythropoiesis, a process involved in recovery from acute anemia. Growing hBMOs in low O2 environments combined with the addition of BMP-4 to media increased the percentage of erythroid cells within organoids by 2.9-fold (Fig. 6B). The changes in erythroid output caused by modifications to these two variables is consistent with a “stress erythropoiesis” phenotype induced by BMP-4 and hypoxia in liquid culture systems.19,20 However, functional testing is needed to confirm this. Evaluating stress erythropoiesis in organoids is a more physiologically relevant human model system which retains the original stem cell populations and is therefore capable of capturing both short- and long-term mechanistic actions of erythroid progenitors during specification, expansion, differentiation, and resolution stages. Combined with gene editing approaches, we can use these modified culture systems to investigate loss-of-function phenotypes, the influence of additional growth factors, the incorporation of other cell types, or how drug treatments may influence human stress erythropoiesis.
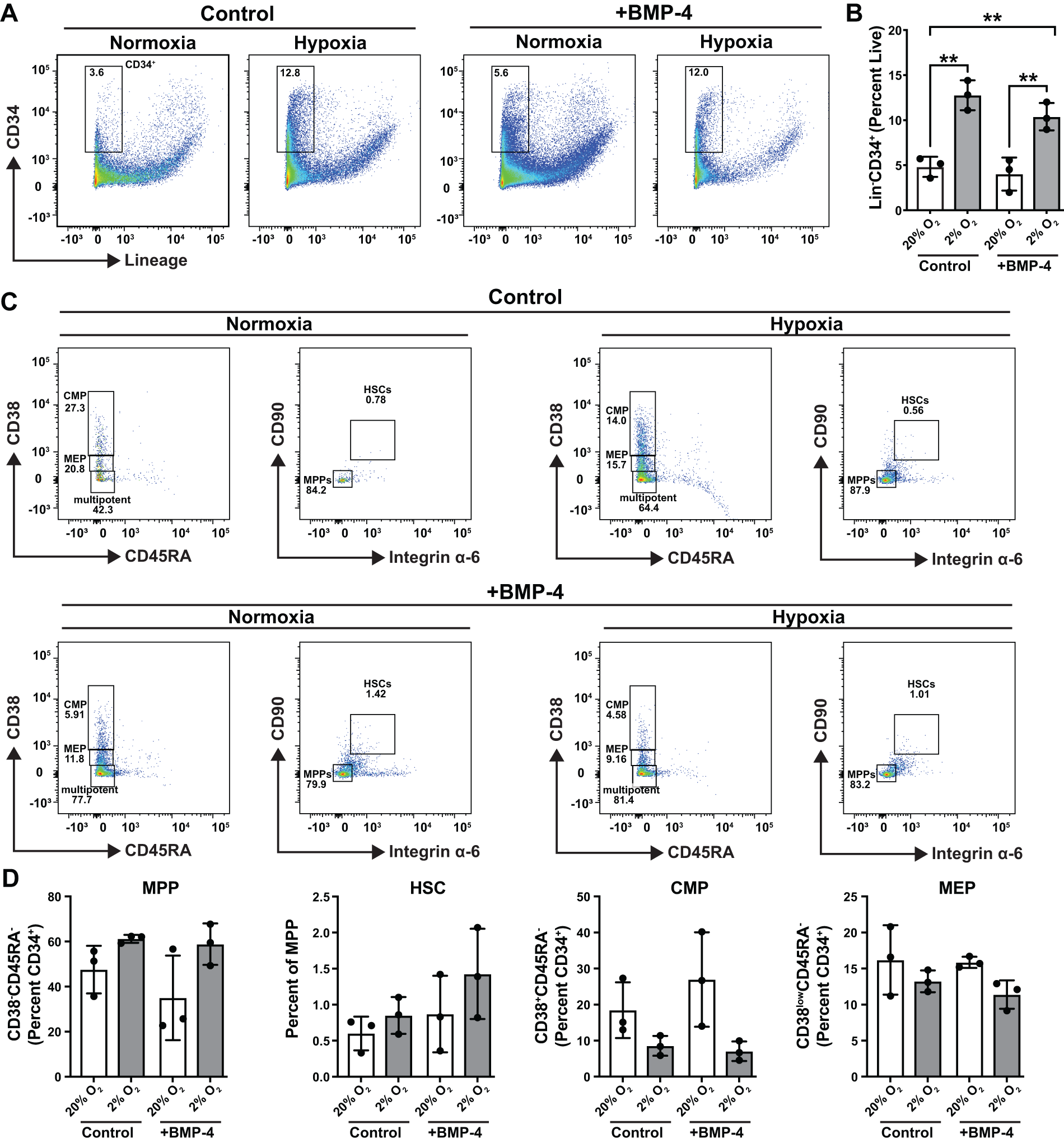
Low O2 and BMP4 alter the HSPC compartment in hBMOs.
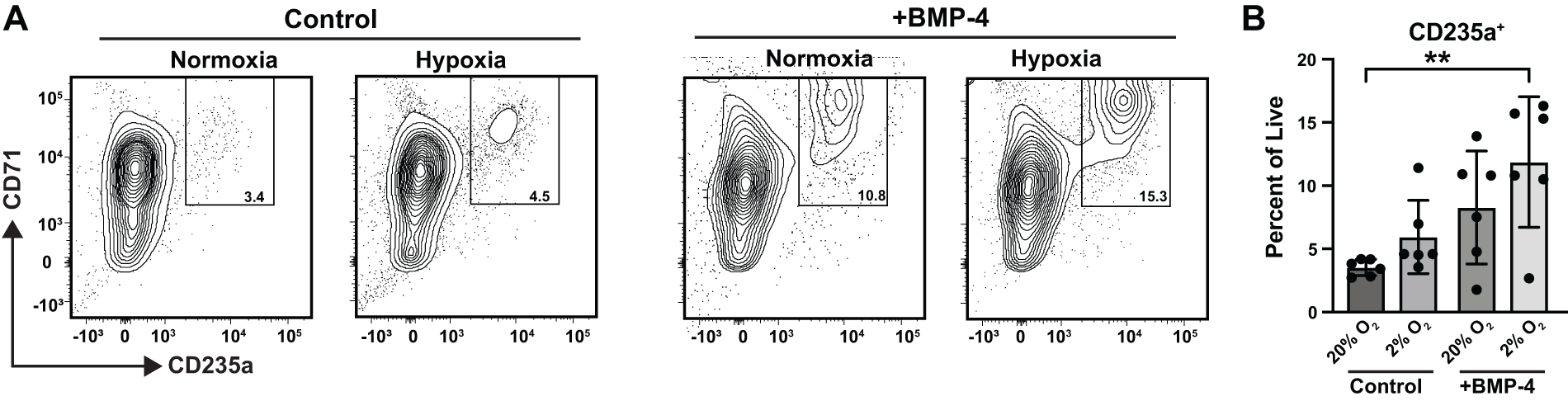
Low O2 and BMP4 increase erythroid output in hBMOs.
Discussion
Good liquid culture protocols are capable of either maintaining hematopoietic progenitor potency, 41 or directed differentiation to specific lineages.42,43 However, these protocols cannot do both and cannot model the multilineage differentiation that occurs in vivo from single progenitors while maintaining progenitor pools. In this lens, hBMOs present significant improvements over liquid culture systems for interrogating mechanisms of human hematopoiesis. Building on prior model systems, which used feeder cells and/or engineered scaffolds,44–46 current protocols have improved experimental strategies to mimic normal human bone marrow architecture.33–35 Since organoid environments mirror the native architecture of stem cell niches, normal hematopoiesis can ideally occur with little input. Paracrine signaling among multiple cell types within the niche permits HSPC multilineage differentiation that cannot be accurately reproduced with liquid culture differentiation protocols. Significant advancements in disease modeling are also possible using hBMO approaches, since the transformation events involve their interaction with native niche environments. Recently, disease models using organoids have adapted organoids to study a range of hematological conditions, including multiple myeloma, 47 MDSs, 48 and myelofibrosis. 33 Since hBMOs arise from iPSC lines, one can envision the development of personalized incubators for drug screening in hematological disease.
For the first time, our study tests applications for the hBMO system using gene-edited CD34+ HSPCs engrafted into organoids. Gene-edited cells were able to maintain a population of immunophenotypically defined HSCs and progenitors and retain their differentiation capacity. Gene editing of primary donor-derived cells can be difficult to quantify with conventional sequencing tools. Since the SAMD1 gene is GC-rich and prone to sequencing and PCR errors, our study required the development of a qPCR-based approach (described in methods) that may prove useful at other GC-rich gene loci for determining editing frequency. As one caveat of our data, we did not conduct parallel functional assays in colonies or mice to evaluate HSC functions in engrafted cells. In addition, while CFSE staining is a robust readout by flow cytometry, we could not distinguish this marker from background after fixation, suggesting that more robust genetic labeling may be required for visualizing engrafted cells in the bone marrow niche. In hBMO-integrated loss-of-function engraftment approaches to test gene activities, FACS purification and functional assays will need to be incorporated into the protocol. Using these approaches, we can define the functional requirements of specific genes along the gamut of human hematopoiesis. For example, while the GATA2 transcription factor is required for human and mouse HSC functions, its expression dynamics, cis-regulatory control, and dosage sensitivity diverge across species.49–51 Human GATA2 haploinsufficiency and regulatory mutations cause bone marrow failure and leukemia predisposition phenotypes that are incompletely modeled in murine systems.49,51 The development of hBMO systems opens the door to examine multiple aspects of haploinsufficient genetic diseases in hematopoiesis, including their roles in HSPC maintenance, hematopoietic differentiation and disease, and influence on bone marrow architecture.
For the first time, we also describe a method for inducing “stress erythropoiesis” in hBMOs. Human stress erythropoiesis has been previously studied in liquid culture assays that have been extremely useful for time course experiments and testing mechanistic requirements for recovery in acute anemia. 21 In our experiments, exposure to low oxygen resulted in expansion of CD34+ cells in hBMOs, and higher concentrations of the BMP-4 growth factor in media increased the output of erythroid populations in combination with low oxygen, demonstrating that hBMOs are responsive to external stimuli needed to initiate regeneration in acute anemia. The response mimics what happens within in vitro systems, while also retaining the HSC pool that would be responsive to subsequent insults or sensitive to genetic perturbation. 21 For these reasons, hBMOs are a significant upgrade as a model system for erythroid stress. Since hBMOs contain more cellular diversity and niche-derived signaling inputs, this provides a physiologically relevant platform to capture additional defining features of stress-responsive erythroid programs that cannot be evaluated in liquid culture. Mimicking physiological stress in hBMOs opens new avenues to study anemia pathogenesis and niche regulation using functional assays, single-cell transcriptomics and other molecular/functional profiling approaches.
Further refinements to hBMO protocols will integrate additional cells and architectural features in human bone marrow. For example, the hBMO protocol used in our study does not support or produce T cells or B cells. 33 The presence of mature lymphoid cells is important even for investigations into myeloid or erythroid cells, since they contribute to HSPC activity.52,53 Lymphoid potential can be stimulated during iPSC differentiation using Wnt and Activin/Nodal inhibition. 35 The two protocols are quite similar, since the initial steps include mesodermal commitment of aggregates, followed by hemogenic endothelium priming, and similar growth factor cocktails and timings are used. Both studies conducted extensive molecular, cellular, and functional phenotyping. In both the Khan et al. (used here) and Frenz-Wiessner et al. protocols, there are limitations, including the numbers of functional HSCs, heterogeneity between different individual organoids, and fetal-like origin based on profiling and transcriptomics. Ongoing work continues to evaluate the fidelity of hBMOs to model human hematopoiesis and disease states.
Supplemental Material
sj-docx-1-ten-10.1177_19373384261463760 — Supplemental material for Applications of the Human Bone Marrow Organoid System to Study Hematopoiesis and Anemia Stress
Supplemental material, sj-docx-1-ten-10.1177_19373384261463760 for Applications of the Human Bone Marrow Organoid System to Study Hematopoiesis and Anemia Stress by Meg A. Schaefer, Adrian R. Black, R. Katherine Hyde, Daniel A. Kuppers, and Kyle J. Hewitt
Supplemental Material
sj-xlsx-2-ten-10.1177_19373384261463760 — Supplemental material for Applications of the Human Bone Marrow Organoid System to Study Hematopoiesis and Anemia Stress
Supplemental material, sj-xlsx-2-ten-10.1177_19373384261463760 for Applications of the Human Bone Marrow Organoid System to Study Hematopoiesis and Anemia Stress by Meg A. Schaefer, Adrian R. Black, R. Katherine Hyde, Daniel A. Kuppers, and Kyle J. Hewitt
Footnotes
Acknowledgments
The authors are grateful for the core facilities at University of Nebraska Medical Center (UNMC), most notably the Organoid Core as a component of the Nebraska Center for Molecular Target Discovery and Development; and The Flow Cytometry Research Facility, which is supported by the Nebraska Research Initiative; and The Fred and Pamela Buffett Cancer Center. We acknowledge the support from the National Institutes of Health, the Nebraska Center for Molecular Target Discovery and Development, and the Nebraska Research Network in Functional Genomics and an F31 predoctoral fellowship for M.A.S. The CD34+ CRISPR editing work was made possible by the Hematopoietic Progenitor/Stem Cell (HPSC) Gene Editing core of the Fred Hutchinson Cancer Center. These contents are the sole responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health (NIH).
Data Availability
Data are available upon request.
Author Disclosure Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding Information
This study was supported by a Team Science Supplement Award to the Nebraska Research Network in Functional Genomics (
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.