
Abstract
Skeletal muscle tissue engineering (SMTE) is a rapidly evolving field with applications spanning regenerative medicine, disease modeling, drug screening, and biohybrid robotics. Effective SMTE requires scaffolds that reproduce the anisotropic architecture and mechanical properties of native muscle while supporting macroscale tissue formation. Decellularized tissues are strong candidates; however, existing approaches face key limitations. Whole-muscle decellularization requires complex perfusion systems and often fails to fully clear the tissue core. In contrast, minced-tissue processing completely disrupts native architecture and necessitates technically demanding reconstruction. As a result, producing long, continuous scaffolds needed to model or restore physiologically relevant muscle units remains challenging. Here, we present an intermediate strategy that enables the fabrication of long, aligned scaffolds from native muscle bundles. Bundles exceeding 10 cm in length were dissected to preserve native alignment and subjected to mild detergent-based decellularization, achieving efficient removal of cellular material while maintaining extracellular matrix structure and mechanics. The resulting scaffolds supported myogenic cell adhesion, proliferation, and differentiation, demonstrating their suitability for in vitro muscle tissue culture. This accessible approach provides a straightforward route to generate macroscale, structurally faithful skeletal muscle scaffolds, bridging the gap between whole-muscle and minced-tissue decellularization methods.
Impact Statement
Current skeletal muscle tissue engineering lacks an accessible way to generate long, aligned scaffolds that accurately reproduce native architecture, limiting both physiological modeling and regenerative applications. Our work introduces a simple strategy that preserves native bundle alignment while avoiding both the complexity of whole-muscle perfusion and the architectural loss associated with minced-tissue methods. This approach enables the routine fabrication of centimeter-scale, structurally faithful scaffolds that support myogenic culture and mechanical conditioning. By making physiologically relevant muscle constructs more feasible to produce, this method expands opportunities in regenerative medicine, disease modeling, and biohybrid system development.
Introduction
Skeletal muscle tissue engineering (SMTE) is a rapidly evolving field with diverse interdisciplinary applications, spanning from regenerative medicine to biorobotics. 1 In the biomedical domain, engineered muscle constructs are being developed for tissue grafting,2,3 disease modeling, and drug screening, 4 offering controlled environments to study muscle development and physiology. Simultaneously, SMTE advances inform the development of biohybrid systems, 5 where muscle-inspired constructs are integrated into robotic platforms to enable biologically inspired movement. This broad range of desired applications highlights the high potential of SMTE, underscoring the importance of developing physiologically relevant muscle constructs suitable for both biomedical and biohybrid applications. Yet, despite significant recent progress, reproducing the structural scale and organization of native skeletal muscle remains a major challenge.
SMTE strategies generally rely on the interplay of three key components: a suitable cell source, a scaffold that guides tissue organization, and biochemical or mechanical cues that promote maturation.6,7 Among these, the scaffold plays a crucial role. Its physical and biochemical properties provide structural support for three-dimensional (3D) tissue formation 8 and strongly influence cellular behavior. 9 To be suitable for SMTE, a scaffold must possess properties tailored to the specificities of skeletal muscle and should therefore: (1) be biocompatible, (2) possess sufficient mechanical strength to withstand the cyclic strains used in muscle maturation protocols, (3) mimic the anisotropic, hierarchical architectural organization of native muscle, with aligned pores or fibrillar structures, (4) support muscle cells' growth, differentiation, and alignment, (5) be sized to allow myofibers to reach physiologically relevant dimensions, and (6) degrade at a rate compatible with tissue development.1,10
Many natural and synthetic materials have been explored to meet these criteria, 11 but they often fail to capture the biochemical and architectural complexity of the native extracellular matrix (ECM). 1 Consequently, there is growing interest in decellularized ECM scaffolds, which can more faithfully capture some of the native matrix’s features. 12 Multiple decellularization techniques have been investigated for skeletal muscle, falling into three broad categories known as chemical, enzymatic, and physical decellularization.13,14 Each method balances cell removal and ECM preservation differently; however, detergent-based chemical protocols, typically using sodium dodecyl sulfate (SDS), are the most widely employed. 14 Still, identifying an optimal decellularization strategy remains challenging as it must be adapted to tissue geometry. The process must be efficient enough to remove cellular material while being gentle enough to preserve ECM structure and mechanical integrity. 13 Consequently, most existing decellularization methods are applied to small muscle fragments or homogenized tissue, 13 which are subsequently reprocessed using electrospinning, 3D bioprinting, 15 or hydrogel casting. 16 While these approaches offer customizability, they rarely reproduce the highly aligned and hierarchical native architecture of the native muscle ECM. 17 Conversely, whole-muscle decellularization better preserves architecture but often suffers from poor reagent penetration into deeper tissue regions, 8 leaving the tissue core only partially decellularized.
Beyond these biochemical challenges, a major and often overlooked limitation in SMTE is the small size of engineered constructs. Most engineered muscle tissues reported to date measure only millimeters to a few centimeters in length and a few millimeters in thickness,18–39 constrained by diffusion limits and mechanical fragility. While such scales may suffice for many in vitro studies, they may not fully reproduce the mechanical loading environment or long-range fiber alignment of native muscle, in which myotubes can extend up to 10 cm. 40 As a result, current systems may be limited for applications that require macroscale force generation, macroscale contractions, biomimetic strain distribution, or more physiologically relevant dimensions. Extending construct length while maintaining 3D organization would therefore provide an opportunity to better investigate these length-dependent aspects of muscle function and help bridge the gap between microengineered models and larger-scale systems.
In this context, the development of decellularized scaffolds that preserve long, aligned muscle bundles is of particular interest. Based on this rationale, a method is presented here for preparing decellularized skeletal muscle bundles using an approach that lies between whole-organ decellularization and finely minced tissue processing. Intact muscle fibers are isolated through targeted dissection to retain the native matrix architecture, and decellularization conditions are optimized to achieve removal of cellular material while preserving ECM integrity. The resulting scaffolds exceed 10 cm in length, exhibit physiologically relevant anisotropy, and possess sufficient mechanical strength to support downstream handling and stimulation. This work therefore provides a platform to investigate skeletal muscle behavior at extended length scales and contributes toward addressing current dimensional limitations in SMTE.
Methods
Preparation and decellularization of skeletal muscle tissue
Dissection
A 50% (v/v) glycerol buffer was prepared by mixing glycerol (Thermo Fisher Scientific, Waltham, MA, USA) with Milli-Q water containing 50 mM potassium chloride (Thermo Fisher Scientific) and 1 mM Tris(hydroxymethyl)aminomethane (Thermo Fisher Scientific). The pH was adjusted to 7. Fresh psoas major muscles were obtained from a local butcher (Oxford, UK). Muscles were collected from four adult pigs (Sus scrofa domesticus) of typical slaughter weight for commercial meat production. As the tissues were sourced postmortem from a food-grade supplier, no ethical approval was required. The muscles were then thoroughly rinsed with a 1% phosphate-buffered saline (PBS) solution to remove residual blood and debris, submerged in the glycerol buffer, and stored at −20°C overnight. The following day, the glycerol buffer was fully replaced, and the samples were placed back at −20°C for at least 7 days. Subsequently, small bundles of muscle fibers were dissected using fine tweezers and scalpel blades. The resulting muscle bundles, dissected to a length of 10 cm and with an average diameter of 2.5 mm, were stored in fresh glycerol buffer at −20°C until further use.
Decellularization
All procedures were performed under sterile conditions. Dissected muscle fiber bundles underwent four freeze–thaw cycles (−20°C to room temperature) to promote cell lysis. These cycles were performed in glycerol buffer to preserve tissue structure during processing. They were then washed three times with Milli-Q water under agitation at 4°C (30 min, 10 h, 30 min) to remove residual glycerol. Two SDS solutions (0.5% and 1% w/v; Merck, St. Louis, MO, USA) were prepared in Milli-Q water. Washed bundles were incubated in SDS under agitation at 4°C to limit structural degradation during treatment. This was done under one of the four following conditions: 0.5% SDS for 8 h, 0.5% SDS for 24 h, 1% SDS for 8 h, or 1% SDS for 24 h. Samples were randomly assigned to each group. After SDS treatment, samples were rinsed three times in Milli-Q water (30 min, 10 h, 30 min) under similar conditions. Finally, the bundles were transferred to PBS with 1% penicillin–streptomycin (v/v; P/S; Merck) and stored at −20°C until use.
Assessment of decellularization
DNA quantification
Muscle fiber bundles were dried under vacuum at room temperature using a cabinet vacuum desiccator (Lab Companion; SP Industries, Warminster, PA, USA) for 1 h. Dried samples were lysed in 200 µL of 0.2% (v/v) Triton X-100 (Merck) in 1× TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) for 30 min. During incubation, samples were vortexed for 10 s every 5 min and kept on ice between vortexing cycles. Lysates were frozen at −70°C overnight, thawed on ice, and assessed for DNA content. To confirm DNA removal following SDS treatment, total DNA was estimated by measuring absorbance at 260 nm (NanoPhotometer NP80; Implen, Munich, Germany). Low DNA samples were then quantified using the Quant-iT™ PicoGreen™ dsDNA Assay Kit (Invitrogen, Thermo Fisher Scientific), following the manufacturer’s protocol.
4′,6-Diamidino-2-phenylindole staining
Scaffolds were fixed in 10% formalin (Merck) for 30 min, after which nuclear material was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. Images were acquired using a fluorescence microscope (Eclipse TE300, Nikon, Tokyo, Japan) and processed in Fiji (v2.14.0/1.54f). All images underwent identical processing, namely background subtraction (rolling ball radius = 50 px), and uniform contrast adjustment (max intensity threshold = 180).
Structural analysis
Scanning electron microscopy
Samples were dehydrated through a graded ethanol series (50%, 70%, 90%, and 95%; v/v, Merck) for 10 min each, followed by 100% ethanol for 10 min and then two times for 30 min. Ethanol was then replaced with a 1:1 (v/v) ethanol–bis(trimethylsilyl)amine (HMDS; Merck) solution for 3 min, followed by two 2-min incubations in pure HMDS. After the final incubation, samples were air-dried overnight in a fume hood. Dried specimens were mounted on aluminum stubs using carbon adhesive tape and sputter-coated with gold using a Quorum Q150R coater (Quorum Technologies, Laughton, UK). Imaging was performed using a Zeiss Sigma 300 scanning electron microscope (Carl Zeiss AG, Oberkochen, Germany) at 2 kV electron high tension.
Microcomputed tomography
Scaffolds were freeze-dried for 24 h (VirTis Wizard 2.0; SP Industries) and scanned with a Skyscan 1172 (Bruker, Billerica, MA, USA). The camera resolution was set to an image pixel size of 6.58 μm, and a source voltage of 40 kV was used. Following flat-field correction, a 180° scan was performed with a rotation step of 0.200° and averaging across 10 images. The resulting projections were reconstructed using NRecon (Bruker). Thermal drift was corrected using the least-squares method. Misalignment compensation was tuned to a value of 0.5, while ring artifact reduction was set to 7 and beam-hardening correction to a value of 50%. Thresholding was performed manually for each sample to optimize visualization of muscle fibers.
Histology staining
Scaffolds were fixed in 10% formalin (Merck) for 24 h, embedded in paraffin, and sectioned either longitudinally or transversally. Sections were stained with hematoxylin and eosin (H&E), Alcian Blue, or Sirius Red. Slides were imaged using an Olympus BX40 microscope (Evident Corporation, Tokyo, Japan) and processed with Fiji (v2.14.0/1.54f). All staining steps were performed according to standard histological protocols, and all images underwent color normalization by background division (ImageJ “Calculator Plus,” using a representative background region).
Mechanical testing
Muscle fiber bundles were tested under uniaxial tension in the wet state using an Instron 5582 mechanical testing machine (Instron, Norwood, MA, USA) equipped with a 100 N load cell. Testing was conducted at room temperature in ambient air immediately after removal from PBS. A constant strain rate of 0.1 mm/s was applied until failure. Force–displacement data were recorded and converted to stress–strain curves using the initial gauge length and cross-sectional area. The mechanical parameters extracted from the resulting curves were maximum load, maximum stress, and strain at maximum stress. In addition, the apparent Young’s modulus was determined from the stress–strain curves. Due to the nonlinear (convex) behavior of the samples, two moduli were calculated: (1) a toe-region modulus, defined as the slope of the curve between 0% and 10% strain, and (2) an apparent linear-region modulus, calculated from the higher-strain linear region of the curve, corresponding to between 10% and 30% strain for glycerinated samples and 35% and 60% strain for prepared samples.
In vitro activities of dECM scaffolds
Routine cell culture
Murine myoblast C2C12 cells (ECACC 91031101) were cultured in T75 flasks using high-glucose Dulbecco’s Modified Eagle Medium (DMEM) (4.5 g/L) supplemented with 20% fetal bovine serum (lot 23619; Calibre Scientific, Los Angeles, CA, USA) and 1% P/S (Merck). Cultures were maintained at 37°C, 5% CO2, and 85% humidity. The medium was changed daily, and cells were passaged before reaching 70% confluence.
Scaffold preparation and cell seeding
Decellularized muscle scaffolds were punch-biopsied into segments of 5 mm length to fit 96-well plate wells while retaining their prepared bundle thickness (see the average diameter of prepared scaffolds in Fig. 1E). The scaffolds were then sterilized in 70% ethanol for 30 min, rinsed three times in PBS, and conditioned overnight at 37°C in complete growth medium. C2C12 cells (passage 20) were seeded at 5 × 10³ cells/cm2 in 20 µL medium per scaffold and incubated for 30 min at 37°C to encourage cell attachment. An additional 50 µL of medium was then added to prevent dehydration, followed by a second 30 min incubation. Finally, the volume was brought up to 200 µL per well. After 24 h, the scaffolds were transferred to a new plate to exclude nonadherent cells from downstream analyses. Scaffolds were maintained for 7 days (proliferation phase) with daily medium changes.
Viability and metabolic activity assays
Metabolic activity was quantified on days 1, 3, 5, and 7 using PrestoBlue™ Cell Viability Reagent (Thermo Fisher Scientific) per the manufacturer’s protocol. Fluorescence was measured by a microplate reader at 560/590 nm (excitation/emission). Live/dead staining was performed on days 1 and 7 using calcein-AM and ethidium homodimer-1 (LIVE/DEAD™ Kit, Thermo Fisher Scientific). Images were acquired using a fluorescence microscope (Eclipse TE300, Nikon, Tokyo, Japan) and processed in Fiji (v2.14.0/1.54f). All images underwent identical processing, namely background subtraction (rolling ball radius = 50 px), live/dead channel merging, and uniform contrast adjustment (maximum intensity threshold = 180).
Preliminary differentiation assessment
This experiment was conducted using scaffolds decellularized with 0.5% SDS for 24 h. After the 7-day proliferation phase, the growth medium was replaced with differentiation medium (low-glucose DMEM supplemented with 1% horse serum and 1% P/S; Merck). Scaffolds were cultured with daily medium changes for 5 days. On day 5, scaffolds were rinsed three times with PBS and fixed in 10% formalin (Merck) for 30 min at room temperature. Samples were permeabilized with 0.2% Triton™ X-100 in PBS containing 1% bovine serum albumin (BSA) and blocked with 0.1% Tween-20 in PBS–BSA. Between each step, scaffolds were rinsed three times with PBS. Actin filaments were stained with Alexa Fluor™-conjugated phalloidin, and nuclei were counterstained with DAPI (Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. Fluorescent images were acquired and processed as described above for the live/dead staining.
Degradation study
Scaffold degradation was assessed by monitoring dry weight loss and changes in mechanical properties over time. All experiments were performed using scaffolds decellularized with 0.5% SDS for 24 h.
Mass loss
Scaffolds were freeze-dried for 24 h (VirTis Wizard 2.0; SP Industries) and weighed using an analytical balance (precision 0.1 mg) to determine their initial dry weight. Samples were sterilized in 70% ethanol for 30 min, rinsed twice with serum-free high-glucose DMEM supplemented with 1% P/S, and incubated in the same medium at 37°C under orbital shaking at 160 rpm. At the designated time points (7, 14, 21, and 28 days), scaffolds were retrieved, frozen at −20°C for at least 24 h, and subsequently freeze-dried for 24 h to determine their dry weight. Mass loss was calculated relative to the initial dry weight.
Mechanical properties
A separate set of scaffolds was sterilized, rinsed, and incubated under identical conditions. On days 1, 14, and 28, samples were removed from the medium and subjected to uniaxial tensile testing as described in the “Mechanical Testing” section.
Data analysis and statistics
All experiments were performed with 3–4 independent biological replicates, based on feasibility considerations and in line with common practice in early-stage biomaterials studies. Technical replicates were averaged before analysis. Statistics were performed using GraphPad Prism v.10.3.1. Results are presented as mean ± standard deviation (SD). Comparisons between multiple groups used one-way analysis of variance (ANOVA) with Tukey’s post hoc test to identify pairwise differences. For datasets with small sample sizes (n = 3–4), nonparametric Kruskal–Wallis tests were also conducted to confirm the robustness of the results. Both tests yielded consistent outcomes (e.g., DNA quantification: ANOVA, p = 0.0087; Kruskal–Wallis, p = 0.0058), indicating statistically significant differences among groups regardless of normality assumptions. Repeated-measures data were analyzed using two-way repeated-measures ANOVA with Bonferroni correction. Statistical significance was accepted at p < 0.05.
Experiment
Scaffold preparation
For scaffold preparation (Fig. 1A), whole pig psoas major muscles were incubated in glycerol, dissected into fiber bundles, and finally decellularized by freeze-thawing and SDS treatment, yielding decellularized muscle scaffolds suitable for further characterization. Muscle bundles were reproducibly dissected to lengths exceeding 10 cm while maintaining fiber integrity, as shown by scanning electron microscopy (SEM) imaging of glycerinated tissue prior to decellularization (Fig. 1B). After detergent treatment, scaffolds appeared macroscopically translucent, consistent with cellular removal, while retaining visible overall fiber alignment (Fig. 1C). At higher magnification, SEM imaging confirmed preservation of ECM fiber orientation, resulting in scaffolds with a well-defined anisotropic architecture (Fig. 1D). Higher SDS concentrations caused marked fiber disruption, indicating that SDS concentration was the primary contributor to structural damage over longer incubation times. Scaffold diameter decreased after decellularization under all conditions (Fig. 1E), with 24 h treatments yielding submillimeter scaffolds, a thickness compatible with diffusion-mediated cell survival in the absence of vascular support.41,42
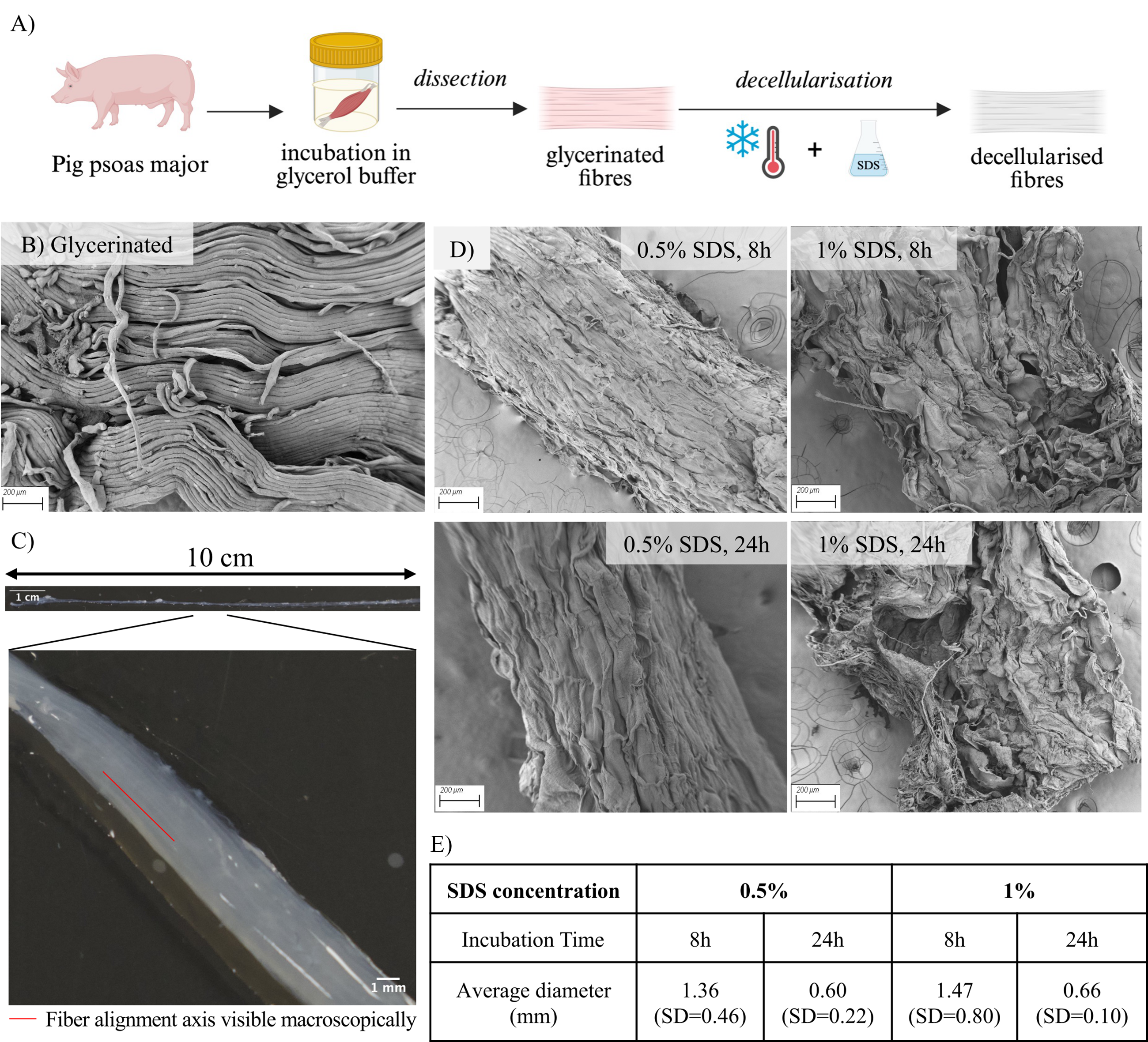
Preparation and preliminary morphological characterization of decellularized skeletal muscle scaffolds.
Assessment of decellularization
DNA quantification confirmed effective decellularization under all conditions (Fig. 2A). Relative to native muscle, DNA content was significantly reduced by 87% following 0.5% SDS treatment for 8 h, by 96% following 0.5% SDS for 24 h, by 90% following 1% SDS for 8 h, and by 92% following 1% SDS for 24 h. PicoGreen analysis yielded absolute dsDNA values of 39.7 ± 9.1 ng/mg, 4.9 ± 4.3 ng/mg, 27.1 ± 11.9 ng/mg, and 4.3 ± 1.9 ng/mg dry weight, respectively (Fig. 2B). All values were below the widely accepted 50 ng dsDNA/mg of ECM dry weight threshold, 43 confirming successful cell removal. DAPI staining and histological assessment provided complementary qualitative information on decellularization efficacy. Native muscle controls showed abundant, well-defined, and aligned nuclei by both DAPI (Fig. 2C, left) and H&E staining (Fig. 2C, right). In comparison, DAPI staining of 8-h-treated samples exhibited only a small number of diffuse, poorly defined signals, consistent with residual nuclear material, whereas no discernible discrete nuclear staining was observed in 24 h scaffolds aside from minor background autofluorescence (Fig. 2D). H&E staining of transverse sections (Fig. 2E) corroborated these findings, showing scattered remaining nuclei following short decellularization and no detectable nuclei after longer treatments. These observations are in agreement with the quantitative PicoGreen results, confirming more complete removal of nuclear material with extended decellularization duration.
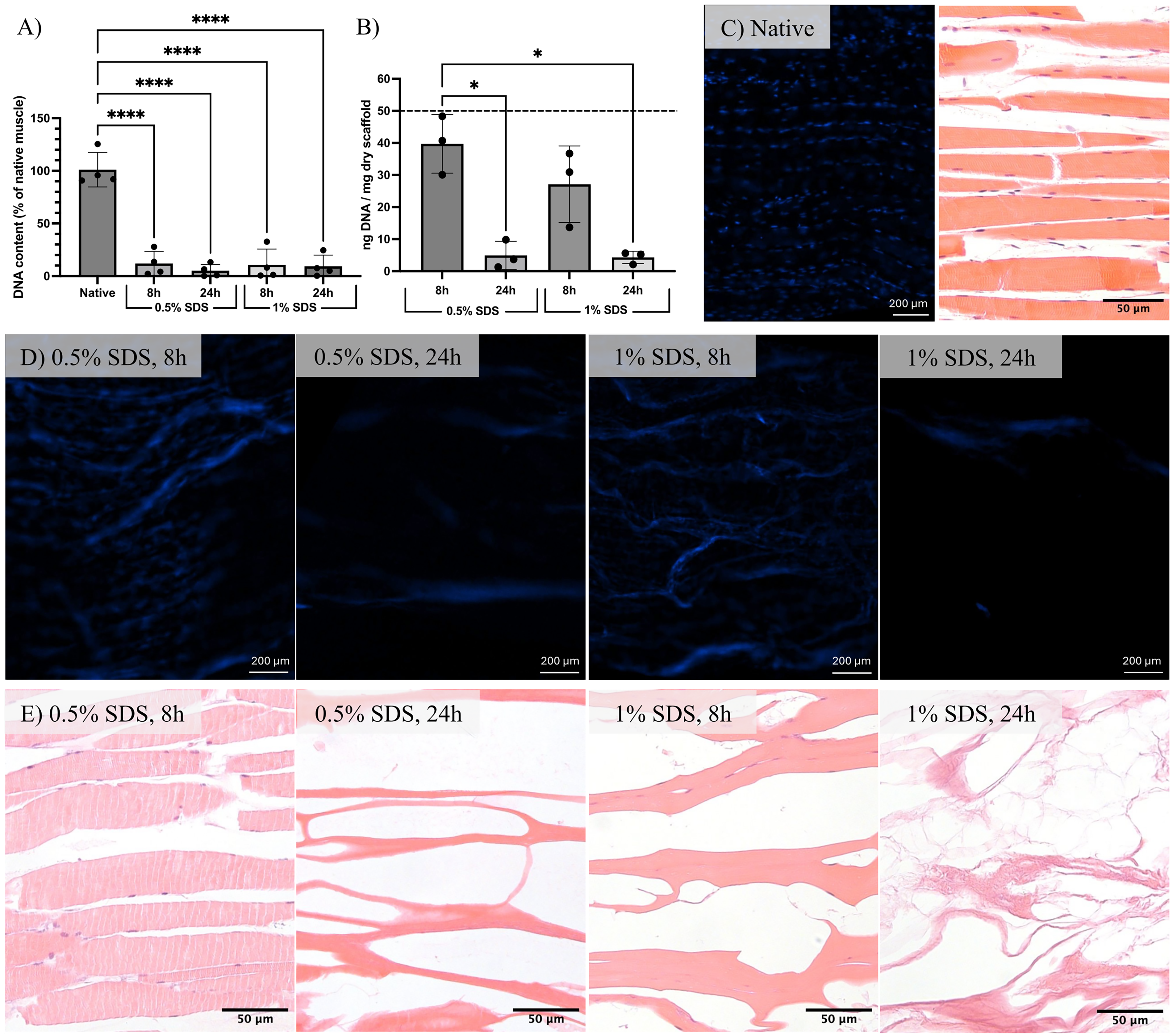
Assessment of scaffold decellularization.
Biochemical, structural, and mechanical characterization
Microcomputed tomography (µCT) imaging of glycerinated controls showed a uniformly dense structure (representative image, Fig. 3A, middle), consistent with the presence of intact cellular and extracellular components. Scaffolds treated with 0.5% SDS for 8 h and 24 h (Fig. 3A, left) exhibited longitudinally aligned tubular voids throughout the construct, indicative of effective cell removal while preserving the native ECM architecture. In contrast, scaffolds treated with 1% SDS showed disruption of structural features. In the 1% SDS for 8 h condition (Fig. 3A, top right), some channels appeared misaligned and folded, as visible in the lower-right region of the image. In the 1% SDS for 24 h condition (Fig. 3A, bottom right), scaffold collapse was observed, consistent with the loss of structural integrity observed in SEM imaging. Across noncollapsed conditions, the mean cavity width was 143 µm. Preservation of structure at lower SDS concentrations was further supported by histological analysis. H&E staining of longitudinal sections from 0.5% SDS for 24 h scaffolds (Fig. 3Bi) revealed the same longitudinally aligned tubular architecture, while transverse sections (Fig. 3Bii) displayed the associated characteristic honeycomb pattern, confirming retention of the native ECM structure. Sirius Red (Fig. 3Biii) and Alcian Blue (Fig. 3Biv) staining of longitudinal sections respectively verified the presence of collagen and glycosaminoglycans (GAGs), consistent with preservation of key ECM components following decellularization. Residual striation patterns were occasionally visible in H&E sections under milder conditions, predominantly in the 0.5% SDS 8 h samples, suggesting that minor amounts of contractile proteins may remain.
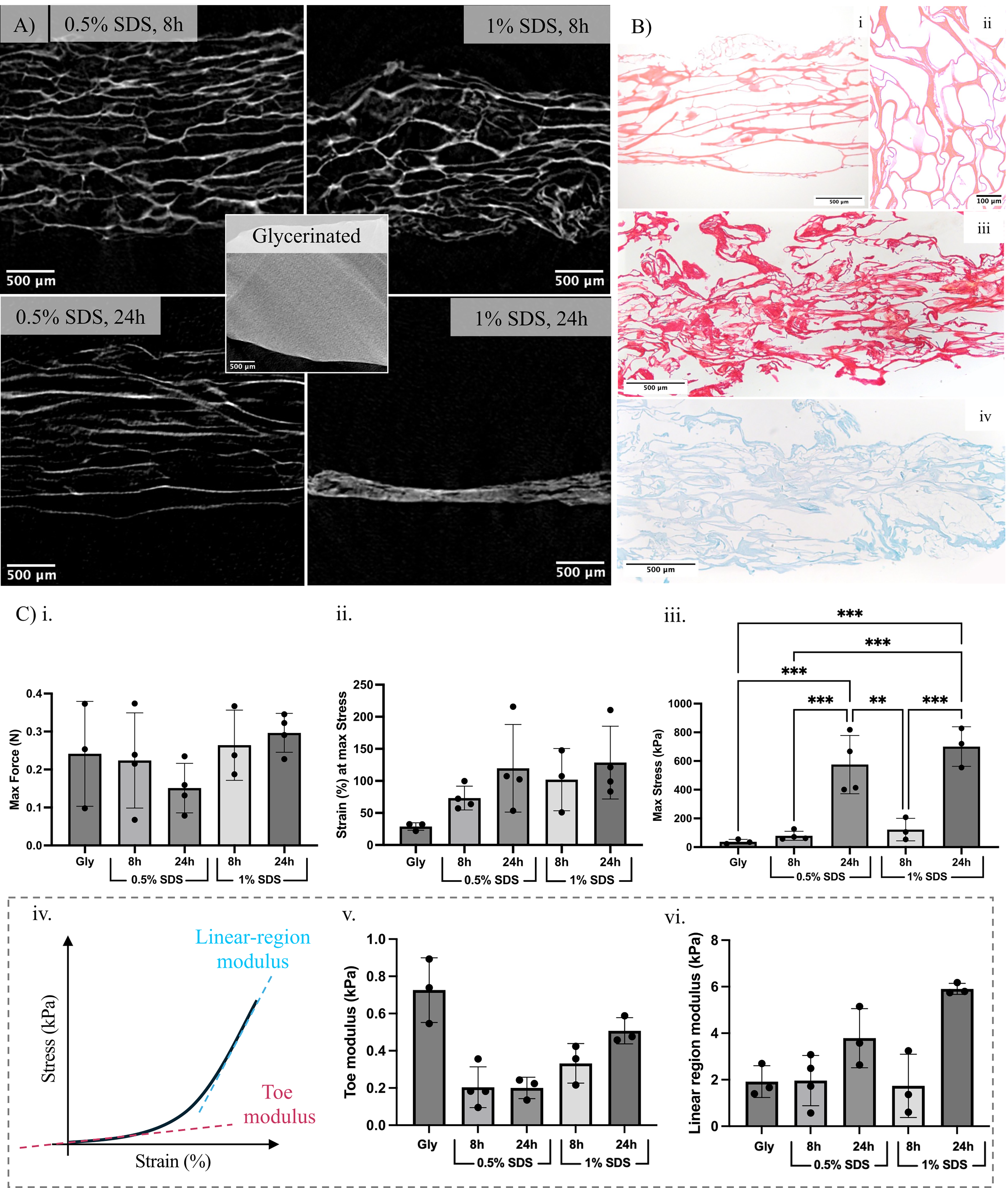
Biochemical, structural and mechanical characterization of decellularized muscle scaffolds.
Mechanical testing demonstrated that decellularized scaffolds retained tensile performance comparable to that of glycerinated controls (Fig. 3Bi–iii), with no significant deterioration across treatments. Maximum force remained stable, indicating preserved load-bearing capacity of the bundles, while increased maximum stress after longer SDS exposure reflected reduced cross-sectional area post-decellularization. Strain at maximum stress was two- to threefold higher in prepared scaffolds compared to glycerinated samples, with higher values observed in more intensively treated scaffolds. However, these differences were not statistically significant. Strain at maximum stress was therefore not considered to differ meaningfully across conditions. Further analysis of the stress–strain curves (Fig. 3Biv) showed that, in the toe region (0–10% strain), decellularized samples exhibited a trend toward lower stiffness compared to glycerinated controls (Fig. 3Bv). In contrast, within the higher-strain apparent linear region, decellularized samples tended to exhibit higher stiffness, with the 24 h samples generally displaying higher modulus values than the other conditions (Fig. 3Bvi). These values fall within the lower range of those usually reported for native skeletal muscle tissue.44,45
In vitro assessment of biocompatibility
Biocompatibility of decellularized scaffolds was evaluated via PrestoBlue metabolic activity assays and live/dead staining. Initial cell attachment to the scaffolds was around 30% for all conditions (34.5 ± 7.3% for 0.5% SDS 8 h, 38.2 ± 16.3% for 0.5% SDS 24 h, 24.6 ± 7.5% for 1% SDS 8 h, 37.0 ± 16.9% for 1% SDS 24 h), as estimated by normalizing the signal from cells on scaffolds to the combined signal from cells on scaffolds and cells remaining attached to the culture wells after transfer of the scaffolds to fresh wells, as described in the Methods section (Supplementary Fig. S1A). Over the subsequent 7 days of culture, C2C12-seeded scaffolds exhibited a progressive increase in metabolic activity, reaching up to 30-fold higher than day 1 levels by day 7 across all scaffold types, with no significant differences between decellularization conditions at any time point (Fig. 4A). Metabolic activity in scaffold-seeded samples remained comparable to that of well controls up to day 5. Data for well controls at day 7 are not shown due to overconfluence and subsequent cell detachment in 2D culture beyond day 5 of culture. Importantly, metabolic activity in all scaffold types was significantly higher than that in unseeded scaffold controls, confirming a negligible contribution from any potential residual cells (Supplementary Fig. S1B).
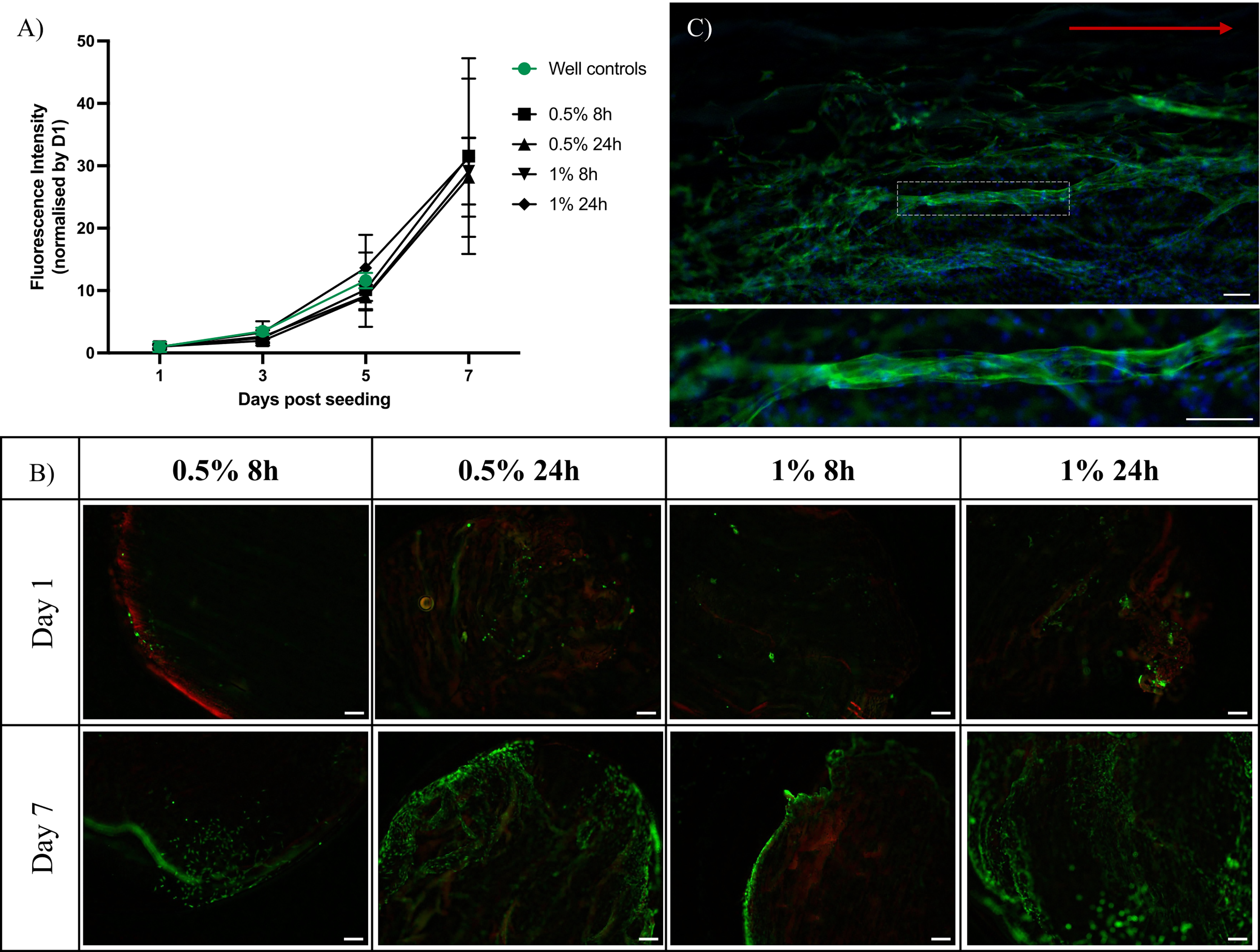
Biocompatibility of decellularized muscle scaffolds.
Live-cell staining corroborated these results (Fig. 4B), revealing predominantly viable cells on all scaffolds at days 1 and 7, with increased cell density over time. Scaffolds exposed to longer SDS incubation appeared to support greater surface coverage and deeper infiltration, suggesting that extended decellularization may enhance scaffold porosity and cell colonization. Complementary dead-cell staining confirmed minimal cytotoxicity, with faint, uniform red signal primarily attributable to scaffold autofluorescence. Collectively, these findings demonstrate that the rise in metabolic activity reflects cell proliferation and infiltration.
Choice of scaffold preparation conditions
To summarize scaffold performance, Table 1 compiles key evaluation criteria encompassing structural, biochemical, mechanical, and biological properties. These criteria were defined as follows: (1) scaffold diameter, with a target of <1 mm to support oxygen and nutrient diffusion; (2) preservation of fiber anisotropy following decellularization, as evaluated by SEM imaging; (3) effective decellularization, defined by DNA content <50 ng/mg ECM and the absence of visible nuclei or nuclear material (H&E, DAPI); (4) porosity, assessed by the presence of µCT-visible channels and evidence of cell penetration into the scaffold interior; (5) mechanical properties, including retention of load-bearing capacity relative to glycerinated controls, strain at maximum stress >40%, and a Young’s modulus compatible with myogenic differentiation; and (6) biocompatibility, evaluated through cell viability and proliferation assays. This overview provides a rationale for selecting the 0.5% SDS for 24 h condition as the most balanced strategy.
Experiments - Choice of Scaffold Preparation Conditions
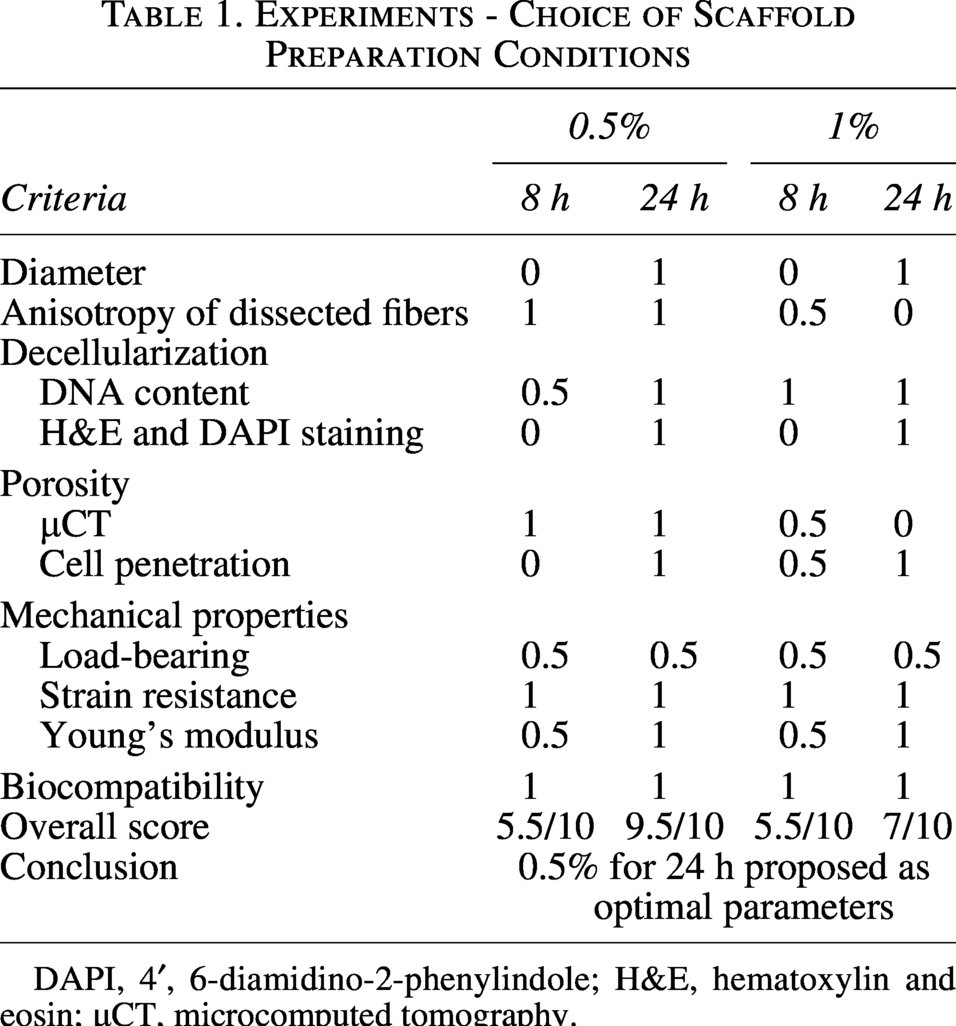
DAPI, 4′, 6-diamidino-2-phenylindole; H&E, hematoxylin and eosin; µCT, microcomputed tomography.
Preliminary assessment of differentiation
Following the proliferation phase described above, myogenic differentiation on 0.5% 24 h scaffolds was assessed after 5 days in differentiation medium (Fig. 4C). Phalloidin/DAPI staining revealed elongated, multinucleated myotube-like structures distributed across the scaffold surface. Myotube structures reached lengths of up to around 1 mm and exhibited general alignment along the principal fiber orientation of the scaffold. These morphological features indicate that the decellularized scaffolds support C2C12 differentiation and promote myotube alignment.
Degradation study
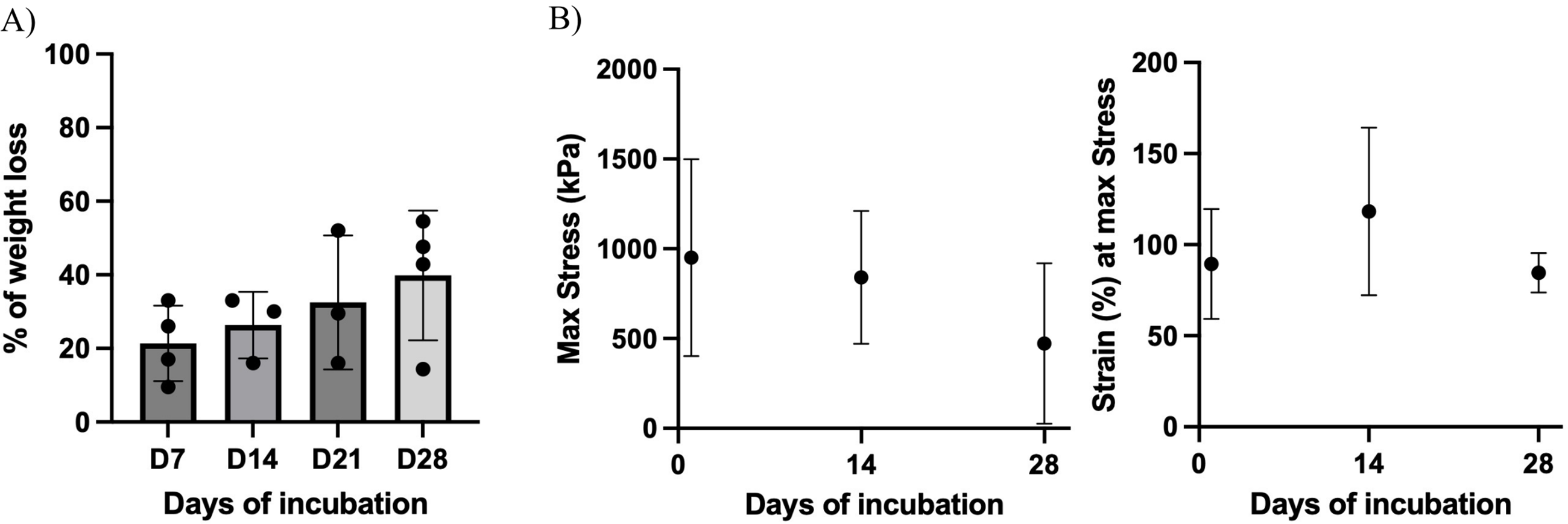
Scaffolds generated using the optimal 0.5% SDS 24 h protocol were then evaluated for degradation over 28 days following incubation in culture medium and freeze-drying (Fig. 5A). Dry weight decreased by ∼20% during the first week (21 ± 10%), with a gradual further decrease to 40% by day 28. However, these changes were not statistically significant across time points. Mechanical testing performed at days 1, 14, and 28 (Fig. 5B) showed a general reduction in maximum stress, roughly halving over the study period, although this trend was not statistically significant. Maximum strain remained unchanged, indicating that the scaffolds largely maintained their mechanical integrity despite modest mass loss and a nonsignificant decrease in strength over time.
Degradation of decellularized muscle scaffolds in vitro.
Discussion
Among scaffold preparation strategies for SMTE, decellularization has emerged as particularly promising. Decellularization can preserve the biochemical complexity of the native ECM 12 and, when applied on whole tissues, it has the additional benefit of retaining the 3D organization characteristic of the tissue. 8 Compared with synthetic or single-component biomaterials, decellularized matrices provide a more physiologically relevant environment for cell adhesion, growth, and differentiation. 1 However, several challenges limit their wider adoption. Key issues include achieving efficient nuclear material removal, maintaining architectural features that support myoblast maturation, attaining physiologically relevant dimensions, and preserving mechanical robustness. Importantly, these requirements are closely interconnected. Harsher decellularization protocols may improve DNA clearance but often compromise ECM structure and mechanics. 13 Reducing scaffold size can facilitate decellularization but requires complex reconstruction to restore functional architecture.13,15,16 Similarly, increasing scaffold dimensions enhances physiological relevance but impedes detergent penetration, heightening the risk of residual DNA being present. 13 Therefore, identifying an appropriate balance between DNA removal, structural preservation, mechanical integrity, and physiologically relevant dimensions remains a major challenge. The work discussed here presents a strategy designed to meet these requirements.
The scaffold preparation strategy developed here was designed to be simple, reproducible, and broadly accessible, relying only on standard laboratory tools and reagents. Although this study was limited by the relatively small number of biological replicates (n = 3–4), which may limit the statistical power to detect subtle differences, consistent trends were observed across independent and complementary assays. In line with this rationale, whole pig psoas major muscles were first incubated in glycerol buffer, then dissected into long fiber bundles and decellularized via freeze–thaw cycles followed by SDS treatment, yielding translucent scaffolds suitable for characterization. The dissection step was central to the method, providing its defining advantage. The goal was to isolate long, thin, parallel-aligned bundles while preserving the native 3D architecture. The psoas major was selected for its favorable anatomy: a fusiform muscle with parallel fibers 46 over 20 cm long and low fat and connective tissue content, making it a reliable source of aligned muscle bundles. Glycerol incubation, as described by Rome, 47 permeabilized membranes, relaxed the tissue, and maintained structural integrity, thereby facilitating clean separation of fiber bundles. 48 Through this approach, using only fine tweezers and scalpels, thin fiber bundles exceeding 10 cm in length could be reproducibly isolated while preserving intact ECM surfaces. Preserving the structure achieved during dissection, including bundle length and ECM integrity, required careful selection of the decellularization protocol. Although a 2% SDS treatment for 24 h detergent-based method was identified as the most widely applied strategy for skeletal muscle, 14 preliminary trials showed that this protocol caused severe damage to the bundles, rendering them unsuitable for further exploration. To minimize disruption, lower SDS concentrations and shorter incubation times were combined with freeze–thaw cycles, which are known to enhance detergent penetration and clearance of cellular debris by disrupting cell membranes through ice-crystal formation. 49 All steps were carried out at low temperature with gentle agitation to preserve ECM proteins and improve detergent diffusion. This strategy yielded translucent scaffolds that retained their bundle length and submillimeter diameter, placing them within the physiologically relevant diffusion limit for oxygen and nutrients and thereby enabling culture without prevascularization. The scaffolds also maintained visible fiber alignment, discernible in the translucent bundles to the naked eye and confirmed by SEM imaging. This anisotropy is critical for guiding muscle cells to align into myotubes capable of coordinated contraction. 50 Notably, the impact of SDS concentration on scaffold integrity was directly observable: Higher concentration visibly disrupted ECM organization, with the most extensive structural damages occurring in scaffolds treated with 1% SDS. These results demonstrate that the combined approach of glycerination, bundle dissection, and decellularization through freeze–thaw cycling and SDS treatment produces scaffolds that are promising candidates for SMTE and warrant further characterization.
Decellularization efficiencie was assessed by quantifying residual DNA, with all conditions yielding values below the accepted threshold of 50 ng/mg ECM dry weight. 43 Treatment duration had a stronger influence than SDS concentration, with 24 h incubations producing the most pronounced DNA reduction. Because residual DNA can trigger inflammatory responses and graft rejection, 51 this enhanced clearance suggests a safer scaffold profile. Consistent with this biochemical assessment, qualitative morphological confirmation of decellularization was provided by complementary histological (H&E) and fluorescence (DAPI) analyses. While scattered or poorly defined residual nuclear material was observed following shorter treatments, improved clearance was observed with extended exposure, even at low SDS concentration. Notably, although many published protocols rely on higher SDS concentrations, harsher detergents, or prolonged perfusion,8,14 these results indicate that milder conditions can achieve comparable outcomes. This may be attributed to a combination of factors, including the prepermeabilization effect of glycerination, 47 as well as the relatively small diameter of the fiber bundles, which likely facilitates reagent penetration and enhances decellularization efficiency. µCT imaging confirmed effective removal of cellular material, revealing empty tubular cavities aligned along the fiber axis. These structures correspond to former myofiber locations and demonstrate preservation of ECM architecture and anisotropy throughout the bundle core. This organization was maintained in scaffolds treated with lower SDS concentrations, whereas higher concentrations led to progressive disruption of structural features and, in the harshest condition (1% SDS 24 h), collapse of the construct, indicating loss of structural integrity, consistent with observations from both SEM and transverse H&E staining. As an aligned tubular architecture provides essential structural cues for myotube alignment and coordinated contractility,10,50 these findings support the use of the less aggressive decellularization conditions. The resulting pore sizes in noncollapsed scaffolds appear suitable for cellular infiltration, with the potential to accommodate a range of relevant cell types, including C2C12 myoblasts (∼25 µm 52 ), which mature into fused myotubes (∼30 µm 53 ) and large human myocytes (∼100 µm 54 ). As skeletal muscle cells are the largest mammalian cells, 55 insufficient pore size is a limitation for achieving deep colonization in SMTE. 10 The large pores observed here should facilitate cell infiltration and broaden applicability across both experimental and translational contexts in SMTE. Alongside the preserved architecture, the biochemical profile of the scaffolds remained consistent with that of native ECM. Collagen and GAG staining were performed to evaluate this as they represent the major structural and biochemical components of native skeletal muscle ECM. 56 Collagen types I and III can account for up to 10% of ECM dry weight 56 and respectively provide tensile strength and elasticity to muscle tissue. 8 Proteoglycans, including GAGs, support matrix hydration 57 and growth factor binding. 56 Both collagen and GAGs were detected throughout the constructs, consistent with previous works on SDS-based muscle decellularization protocols. 57 Together, these results support that the scaffolds retain key structural and biochemical features of native skeletal muscle ECM.
Mechanical testing showed that the scaffolds retained properties relevant to functional muscle constructs. All samples withstood strains exceeding 40%, a level comparable to the upper physiological range of native muscle in vivo and known to promote myotube maturation in vitro.5,58 Maximum force remained unchanged, consistent with preserved ECM integrity. Although scaffolds subjected to longer SDS exposure exhibited higher maximum stress, this reflected scaffold thinning and associated reduced cross-sectional area rather than improved material strength, as overall force capacity was constant. Scaffold thinning in the 24 h samples may therefore reflect higher cellular clearance, consistent with DNA quantification results, which increased porosity while leaving mechanical performance intact. Analysis of stress–strain behavior revealed a nonlinear, J-shaped curve response, in agreement with previous reports for similar biological tissues.59–61 This behavior reflects the characteristic mechanical response of fibrous soft tissues and supports the identification of two distinct regimes, with trends in each region in agreement with prior studies.59–61 In the low-strain (toe) region, decellularized scaffolds exhibited reduced stiffness, likely reflecting increased compliance associated with a more porous and hollow structure, as well as enhanced fiber straightening at initial loading. 60 At higher strains, however, these scaffolds displayed increased stiffness, indicating that once the matrix is engaged, resistance to deformation is preserved or enhanced. The apparent Young’s modulus measured in this higher-strain region, which is the parameter most commonly reported for skeletal muscle, fell within the lower range of values described for native tissue.44,45 Notably, the higher modulus observed in the 24 h condition places these scaffolds within a stiffness range reported to support myogenic differentiation.44,45
Biocompatibility was assessed using PrestoBlue metabolic assays and live/dead staining. Metabolic activity increased across all scaffold types, although this alone cannot confirm biocompatibility as metabolic upregulation is also a hallmark of cellular stress. Live/dead staining provided the necessary confirmation, showing predominantly viable cells with minimal death after seven days. The relatively low number of visible cells in these images, particularly at early time points, is likely influenced by both the initial seeding conditions and imaging limitations. C2C12 cells were seeded at low density, as is appropriate for this cell type, 62 and only ∼30% of the initially seeded cells attached to the scaffold, resulting in few cells detectable at day 1. These seeding efficiencies are consistent with previously reported values for cell attachment to engineered scaffolds, particularly in the absence of dynamic seeding.63,64 In addition, the use of widefield fluorescence microscopy may underestimate cell presence within the 3D scaffold due to limited optical sectioning, potentially obscuring cells located deeper within the material. Despite these limiting factors, a clear increase in live cell numbers between day 1 and day 7 was observed. Together, these results indicate that scaffolds supported cell survival and proliferation, confirming their biocompatibility and validating the decellularization approach. Detergent removal is a key concern in SDS-based methods, since residual detergent can induce cytotoxicity. 65 Here, extended wash cycles proved sufficient to eliminate cytotoxic residues, as evidenced by robust cell growth across all conditions. Interestingly, scaffolds subjected to longer SDS treatments supported greater cell infiltration, likely due to subtle structural changes such as increased porosity or reduced bundle diameter that facilitate cell migration into deeper regions. Achieving deep infiltration is particularly relevant, as limited cell penetration remains a challenge in SMTE. 66 The retained anisotropy of the scaffolds also provided directional cues for cell orientation. By day seven, particularly under the 0.5% SDS 24 h condition, cell alignment with the internal fiber structure was observed. Rather than forming a single continuous alignment, cells organized into local domains broadly oriented in the same direction, though not fully colinear across the scaffold. This pattern likely reflects minor fiber disorganization introduced during handling, but such regional alignment nonetheless represents a positive step toward coordinated myotube formation as even domain-level organization can promote functional maturation. Prestretching or mechanical conditioning before seeding may further enhance global fiber alignment and strengthen the scaffolds’ instructive potential.
Following identification of the 0.5% SDS 24 h protocol as the most promising preparation strategy, a differentiation assay was performed on this condition as a proof-of-concept. The results were consistent with observations from the proliferation stage, with clear cell alignment along the scaffold. The scaffolds supported the formation of elongated, multinucleated myotube-like structures after 5 days in differentiation medium. Phalloidin staining revealed that these cellular structures displayed preferential alignment along the scaffold fiber axis, consistent with the known influence of anisotropic topography on myoblast orientation. 50 While more specific myogenic markers, such as MF20 or tropomyosin, would provide a more comprehensive characterization of differentiation, the morphological features observed here support the conclusion that the scaffold environment permits progression toward a differentiated phenotype. Further quantitative analysis and marker-based validation will be required to fully characterize the extent of myogenic progression.
Altogether, these in vitro assays provide important validation by demonstrating that the scaffolds are noncytotoxic, support cell colonization, and present an architecture conducive to alignment. It should be noted, however, that in vitro compatibility does not guarantee in vivo performance. Factors such as immune response, vascularization, and long-term integration will need to be evaluated before considering clinical applications. Moreover, while this study provides initial qualitative evidence of myotube formation and early differentiation, comprehensive functional assessments, including quantification of fusion, maturation, and contractility, remain to be investigated to determine the scaffolds’ full regenerative potential.
The degradation profile of the selected scaffold was then examined to assess its temporal stability under extended in vitro culture conditions, where maintaining structural integrity over time is essential to properly support cell proliferation, alignment, and maturation. In this context, a 1-month time frame was selected to represent the upper range of durations typically explored in the field, 1 providing a more stringent benchmark for scaffold persistence. Despite modest weight loss, scaffolds maintained their mechanical performance over 1 month, with no significant reduction in maximum forces, stress, or strain. This stability suggests that the preservation of fiber alignment and bundle architecture contributes to maintaining structural integrity and resilience during prolonged incubation. The initial ∼20% dry-mass reduction within the first week likely reflects loss of loosely bound ECM components, after which weight stabilized, suggesting retention of the core fibrillar matrix. The gradual degradation profile observed after this initial drop achieves the desired balance between preserving mechanical competence during early tissue infiltration and permitting eventual resorption.
Together, these findings strengthen the case for the 0.5% SDS 24 h protocol as a robust foundation for SMTE, balancing effective decellularization with preserved architecture, mechanical strength, and stability. Placing these findings in a broader context, this work addressed a key structural bottleneck in the field: the limited size of engineered muscle constructs. By maintaining native fiber alignment over physiologically relevant lengths exceeding 10 cm, these scaffolds overcome the dimensional constraints that have traditionally restricted engineered muscle to millimeter- or centimeter-scale constructs.
Although thickness remains limited by the absence of intrinsic vascularization, the ability to generate long, structurally coherent scaffolds represents a step toward reproducing native muscle structure. Given that force generation in engineered muscle has been shown to scale with myotube length, 67 access to extended fiber dimensions may be relevant for investigating functional performance at more physiologically representative scales. In this context, such constructs open promising avenues for applications including bio-actuated systems and mechanobiological studies that benefit from extended, aligned muscle segments. They also present opportunities for disease modeling in contexts where spatial scale and alignment are important. In particular, these scaffolds may enable investigation of cell behavior over dimensions that more closely approximate native human muscle fibers, which can extend up to multiple centimeters in vivo. 40 This is relevant for studying processes such as myofiber maturation and pathological changes that propagate along the fiber length, as observed in conditions such as muscular dystrophies.68,69 In addition, although the present scaffold format is not intended for direct regenerative use, its geometry may lend itself to modular strategies in which multiple aligned units are combined to generate constructs with increased cross-sectional mass and functional capacity. This approach could provide an alternative route to larger-scale engineered muscle tissues compared with whole-muscle decellularization approaches. Future work will be required to explore these possibilities and determine the extent to which such systems can support functional maturation at larger scales.
Authors’ Contributions
M.Z.C.: Writing—original draft (lead); writing—review and Editing (equal); investigation (lead); conceptualization (equal); formal analysis (lead); and methodology (lead). G.B.: Writing—review and Editing (equal); investigation (supporting); and formal analysis (supporting). S.J.B.S.: Writing—review and editing (equal) and supervision (supporting). P.-A.M.: Writing—review and editing (equal); supervision (lead); conceptualization (equal); and methodology (supporting).
Supplemental Material
sj-docx-1-ten-10.1177_19373384261463762 — Supplemental material for A Method for Fabricating Long Decellularized Scaffolds from Skeletal Muscle
Supplemental material, sj-docx-1-ten-10.1177_19373384261463762 for A Method for Fabricating Long Decellularized Scaffolds from Skeletal Muscle by Maira Z. Cordelle, Giulia Biasi, Sarah J.B. Snelling, and Pierre-Alexis Mouthuy
Footnotes
Acknowledgments
The authors thank Dr. Tania Choreno-Machain for her valuable guidance and assistance during the early stages of laboratory work and training. We are also grateful to Dr. Igor Dyson (Department of Engineering, University of Oxford) for providing access to the Instron system and for his help and guidance with its use. We thank the Histology Service of the Kennedy Institute of Rheumatology (University of Oxford) and more specifically Dr. Ida Parisi for her technical support with histological procedures. We further thank the Center for Human Genetics (University of Oxford) and the Sir William Dunn School of Pathology (University of Oxford) for providing access to their scanners (SEM and µCT, respectively). Figure 1A was created using ![]() .
.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work has been completed with the financial support of the United Kingdom’s Medical Research Council (MRC DTP iCASE 2023 Award), the United Kingdom’s Engineering and Physical Sciences Research Council (rEP/S003509/1), and the UKRI-BBSRC Interdisciplinary Bioscience Doctoral Training Partnership (BB/T008784/1). This project was also supported by the National Institute for Health and Care Research Oxford Biomedical Research Centre (NIHR203311), and the EPA Cephalosporin Graduate Scholarship (Charity number: 309698).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.