
Abstract

Keywords
A previous healthy 11-year-old child with a 24-hour history of asthenia, diplopia, dysarthria, dysphonia, swallowing difficulty, and vomiting was admitted to our institution on Christmas eve. The child complained of perioral paresthesias and blurry vision 2 days prior to admission and an unusual tiredness after skiing on the day of admission.
He presented with a temperature of 36.8°C, blood pressure 100/70 mm Hg, pulse 80 beats/min, respiratory rate 20/min, and oxygen saturation 99% on room air. He was awake, well oriented and his physical examination was within normal limits. Neurological examination showed bilateral ophthalmoplegia with unreactive pupils, dysphonia, dysphagia with minimal limb weakness with associated hyporeflexia. Sensation was maintained, but he presented with urinary retention described as a poor urinary stream with intermittent flow, straining, and a sense of incomplete voiding and hesitancy. A complete blood count, electrolytes, arterial blood gas, C-reactive protein and herpes virus 1 and 2 (HSV-1 and HSV-2), Epstein-Barr virus (EBV) antibodies were within the normal range. Cerebrospinal fluid (CSF) examination was unremarkable (glucose 81 mg/dL, normal values 40-70), protein 20 mg/dL (normal values 15-45), lactate 11.7 mg/dL (normal values 10.8-19.8), and no erythrocytes or leucocytes. Polymerase chain reaction (PCR) for neurotropic viruses (human herpes virus 6, human herpes virus 7, human herpes virus 8, adenovirus, Parvovirus, EBarrV, herpes simplex virus 1 and 2, and varicella-zoster virus), cultures for meningococcus and fungi, and serology for Borrelia, Chlamydia Pneumoniae, enterovirus, influenza A and B, metapneumovirus, morbillivirus, and Mycoplasma Pneumoniae were all negative. The electroencephalography (EEG) was also normal. Intoxication or snakebite were excluded by medical history, a detailed neurological examination, and laboratory tests (urine and plasma toxicology screen for amphetamines, cannabinoids, cocaine, opiates, phencyclidine,alcohol, benzodiazepines and tricyclic antidepressants). There was no history of ingestion of homemade or rotten food that could contain botulinum spore or toxins. Twenty-four hours after admission, the patient developed respiratory failure and areflexia of the upper limbs requiring mechanical ventilation and admission to the pediatric intensive care unit (PICU). Repeat CSF examination showed normal glucose, protein and lactic acid, negative bacterial cultures, and PCR for viruses. No albuminocytologic dissociation was observed.
Under the working diagnosis of a Miller-Fisher syndrome (MFS), the child was treated with intravenous immunoglobulin G (IgG) infusion (400 mg/kg) for 5 days with no clinical response, followed by 5 cycles of plasmapheresis. The child gradually recovered and it was possible to perform electromyography and nerve conduction studies (EMG/NCSs) which showed a normal conduction pattern. The edrophonium test, antibodies to voltage-gated calcium channels, to acetylcholine receptors (AChRs), and anti-ganglioside autoantibodies (IgG and IgM for GM1, GM2, GD1a, GD1b, and GQ1B) were also negative. A diagnostic result was received.
Descending Cranial Nerve Palsy in Pediatric Age
Discussant: P. E. Cogo
A standard evaluation for descending cranial nerve palsy usually includes EMG and NCSs and CSF analysis and neuroimaging to detect the underlying causes. In addition to intoxication, viral or bacterial infections and the presence of autoantibodies in blood, stool, and CSF have to be ruled out. In pediatric patients, special attention is always given to inherited disease, although in this adolescent patient many congenital or early onset diseases need not be considered. Acquired myasthenic syndromes represent a broad spectrum of different pathologies that can affect both pre- and postsynaptic neurons. Myasthenia gravis (MG) and the Lambert-Eaton myasthenic syndrome (LEMS) are both acquired autoimmune disorders characterized by a defective neuromuscular transmission. Several clinical features help distinguish between MG and LEMS, such as decreased tendon reflexes and autonomic dysfunction that are unique to the latter. 1 In both these conditions, the detection of autoantibodies is one of the most important steps for the diagnosis. Seropositive (SP) MG includes disease with circulating antibodies to the AChR, while seronegative (SN) patients lack these antibodies. Recently, antibodies to muscle-specific tyrosine kinase (MuSK) have been demonstrated in over 40% of patients with generalized SN MG. 2 –5
The LEMS is an autoimmune disorder usually associated with autoantibodies against the presynaptic voltage-gated calcium channel, which leads to neuromuscular blockade. 6 The test for antibodies against the AChRs can also be positive in some cases. While the LEMS can be found as a solitary disease, 60% of cases have an associated small cell lung cancer (40% even before the first symptoms appear). Patients may also be diagnosed with other neoplasms. Electrophysiological examination can be helpful because a decrement in compound muscle action potential (CMAP) amplitude with high-frequency stimulation confirms the diagnosis. Corticosteroids, azathioprine, and 3.4-diamidopyridin are used to treat LEMS, while acetylcholinesterase inhibitors are less efficient. Plasma exchange and intravenous immunoglobulins (IVIGs) may be effective in some patients with LEMS. 7
Juvenile MG is an autoimmune disorder, while congenital MG results from genetic mutations that impair neuromuscular transmission and they are both very rare. 8 There are also some rare form of congenital MG recently described, which can first become evident during adolescence or even adult life, for example the slow-channel syndrome. 9,10
Organophosphate intoxication causes inhibition of acetylcholinesterase in the central and peripheral nervous system resulting in an accumulation of acetylcholine at the neuromuscular junction. 11 Together with flaccid paralysis, seizures are a typical feature of organophosphate intoxication in children. Since these can rapidly progress to status epilepticus, contributing to neuronal damage and neurological impairment, EEG monitoring is mandatory when intoxication is confirmed. 12
Miller-Fisher syndrome is an uncommon variant of Guillain-Barré syndrome, an acute inflammatory polyneuropathy most commonly characterized by rapidly progressive, essentially symmetric weakness, and areflexia, occurring both in adults and in children. 13 Miller-Fisher syndrome is known to be rare in children (0.09/100 000) and is characterized by ataxia, ophthalmoplegia, and progressive weakness of the upper limbs. 14
Despite the near eradication of poliomyelitis, acute flaccid paralysis caused by viruses remains a major central nervous system complication associated with several infections as West Nile virus (WNV) and tuberculosis. Poliovirus (PV) is the causal agent of paralytic poliomyelitis resulting in flaccid paralysis in around 1% of infections. This is usually asymmetric with fever, headache, nausea, vomiting, and pleocytosis characteristic of exordium that are absent in MFS. The maximum extent of the paralysis is reached in 3 to 4 days, definitive laboratory diagnosis requires isolation of PV from stool samples, CSF, or oropharyngeal secretions in cell culture systems of human or monkey origins (primates cells). Other enterovirus (types 70 and 71), echoviruses, and coxackieviruses can cause an illness-simulating PV. 15,16 Also patients infected with WNV may develop acute neurologic disease, which can be severe or even fatal, including meningitis, encephalitis, and an irreversible acute flaccid paralysis or poliomyelitis-like syndrome. 17 Diagnosis depends on the demonstration of WN-specific IgM antibodies in CSF that usually shows a pleocytosis, with a predominance of neutrophils in half the patients. Neurophysiological, radiological, and pathological studies suggest WNV causes acute flaccid paralysis by damaging the anterior horn cells in the spinal cord. The clinical presentation is probably best described as a “poliomyelitis-like illness,” but other findings during the recent outbreaks include increasing the recognition of a Parkinson disease like presentation and movement disorders. 18
Tuberculous radiculomyelitis (TBRM) is a complication of tuberculous meningitis (TBM), which has been reported rarely in the modern medical literature and especially in immunosuppressed patients. The most common symptoms are subacute paraparesis, radicular pain, urinary retention, and subsequent paralysis. In most patients with TBRM, evaluation of CSF reveals an active inflammatory response with pleocytosis (lymphocytosis), hypoglycorrhachia, and a very high protein level. Magnetic resonance imaging (MRI) and computed tomography (CT) scans are critical for diagnosis, revealing loculation and obliteration of the subarachnoid space along with linear intradural enhancement. 19
Detailed collection of clinical symptoms in this patient represented a key point in identifying the possible etiology of his descending cranial nerve palsy. Important considerations were (1) the lack of any family history for neurological or autoimmune disorders, especially for inherited neuromuscular disorders such as MG; (2) the acute onset and progression of the disease over a short period of time, without the associated cognitive or behavioral symptoms; (3) no history of recent medications or ingestions (organophosphates, drugs or rotten or homemade food). Meningitis was ruled out with the spinal fluid examination showing a normal result. Table 1 lists the differential diagnosis supported by the clinical presentation.
Differential Diagnosis for a Patient With Cranial Nerve Palsies and Descending Weakness
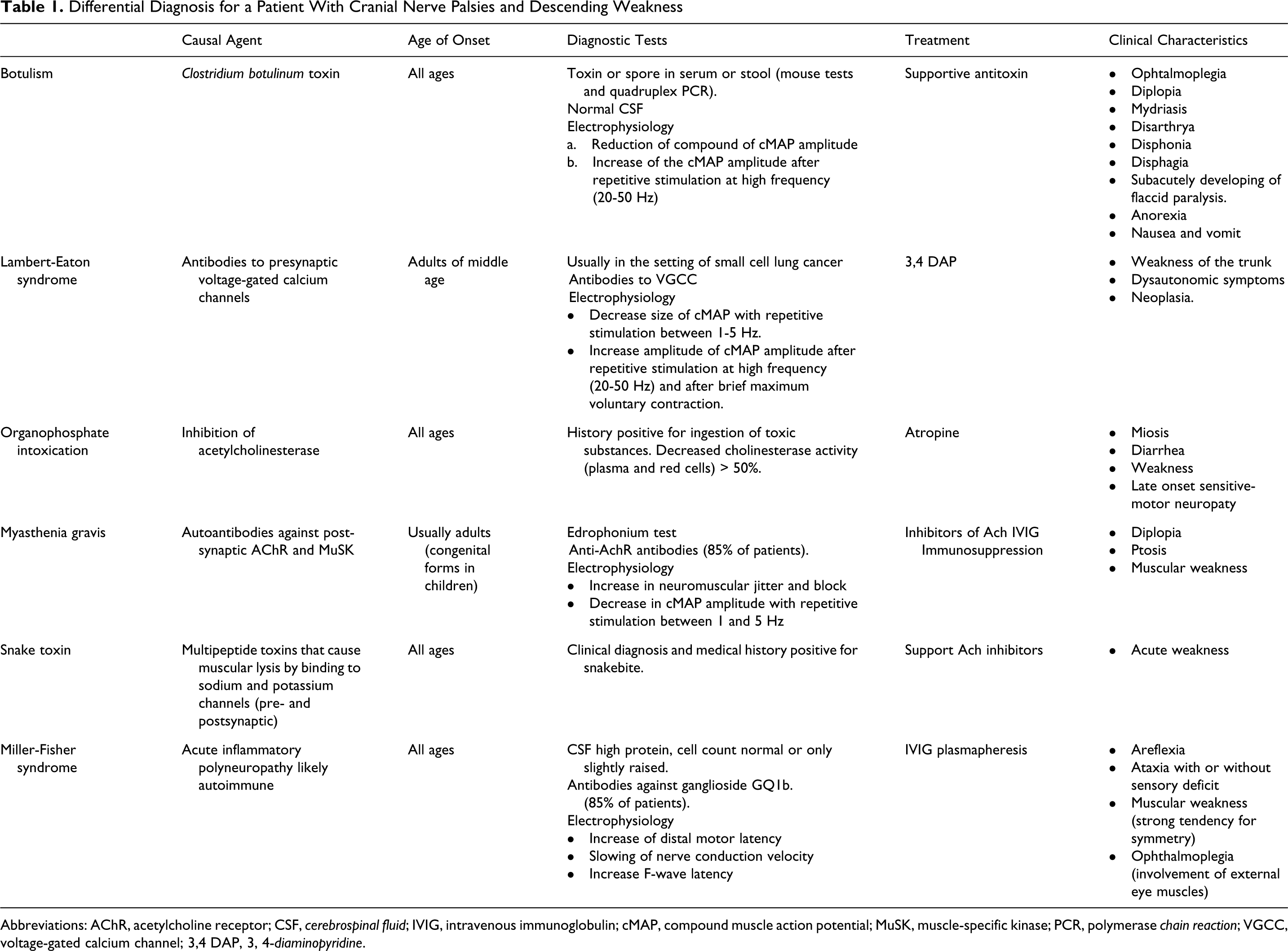
Abbreviations: AChR, acetylcholine receptor; CSF, cerebrospinal fluid; IVIG, intravenous immunoglobulin; cMAP, compound muscle action potential; MuSK, muscle-specific kinase; PCR, polymerase chain reaction; VGCC, voltage-gated calcium channel; 3,4 DAP, 3, 4-diaminopyridine.
After the first examination, MFS was considered as the most probable cause of this clinical exordium.
Diagnostic Result
Normal EMG/NCS and the absence of IgG antibodies against the ganglioside GQ1b made the diagnosis of MFS questionable and led us to pursue tests for botulism even though the child did not report possible ingestion of spores or toxins and did not live near a construction site that could release spores. A serum sample and a fecal swab were collected 1 month after the clinical exordium and submitted for botulinum neurotoxins (BoNTs), and BoNT producing clostridia detection at the National Reference Center for Botulism (NRCB) but both were negative. 20 Considering that these samples were obtained late in the disease course and after the patient had undergone plasma exchange we decided to send another serum sample collected on Christmas day during the first hospitalization. Botulinum toxin was detected by Mouse bioassay (Centers For Disease Control and Prevention method) and the diagnosis of botulism was confirmed.
The patient showed progressive neurological improvement after 4 and 6 weeks and complete recovery at 12 weeks from the disease onset.
Botulism
Discussant: L. Fenicia
Botulism is a paralytic illness caused by BoNT produced by the anaerobic spore-forming bacterium Clostridium botulinum and rarely by C baratii and C butyricum. 21
It is characterized by cranial nerve palsies and a descending flaccid paralysis, which can involve respiratory muscles, leading to neuromuscular respiratory failure. Paralysis is a consequence of the neurotropic action of the toxin that prevents fusion of synaptic vesicles containing acetylcholine at the nerve terminal. This inhibits the release of acetylcholine at the neuromuscular junction causing symptoms ranging from mild hypotonia to severe, flaccid paralysis. This descending flaccid paralysis, unless treated immediately with supportive therapy, can be fatal. The differential diagnosis of this condition is complicated and includes common causes of hypotonia such as sepsis or meningitis, as well as more uncommon disorders that are not easy to reveal in an emergency context. 22 For this characteristic of being a life-threatening condition, botulism often represents a difficult clinical challenge especially where a rapid diagnosis is essential.
Five different forms of the illness are described 23 :
Foodborne botulism is caused by ingestion of food contaminated with botulinum toxin.
Iatrogenic botulism is caused by incorrect use of the toxin for cosmetic or therapeutic aims.
Wound botulism is caused by C botulinum colonization and in situ toxin production.
Infant botulism affects children younger than 1 year of age and is caused by the absorption of the BoNT produced by clostridia that temporarily colonize the intestinal tract.
Adult intestinal toxemia botulism is a rare form of intestinal colonization and toxin production in the older child or adult.
The majority of the literature describes the epidemiology of infant botulism 23,24 that usually occurs in the first 6 months of life. Regarding intestinal toxemia botulism in adults outside the United Sttaes, about 20 cases have been reported worldwide mainly related to risk factors such as surgical interventions, immune system disruption, and prolonged antibiotic therapy. In Italy, 4 cases have been diagnosed, 21,25 with these singular characteristics and in particular 2 of them were young and previously healthy people: a boy (9 years) and a girl (19 years). Curiously these patients also fell ill during the Christmas holidays in 2 consecutive years and a rare strain of type E C butyricum was isolated in both cases from fecal samples. Hospitalized for atypical abdominal pain, both patients underwent a laparotomy and resection of a Meckel diverticulum.
In this case although the symptoms were strongly suggestive of botulism, the age of the patient was confounding: he was too old to present with infant botulism and considering his recent history the foodborne form was unlikely. In this situation, the possibility of intestinal colonization has to be considered.
The diagnosis of botulism can be very difficult especially in an emergency context. Botulism is suspected on clinical grounds and confirmed by laboratory investigation and should always be considered in the presence of a descending cranial nerve palsy, even if the clinical history raises no suspicion for the ingestion of toxins.
Diagnosis of botulism is based on the identification of toxins or spore in serum or stool. It is also prudent to analyze the available biologic, environmental, and alimental samples that are suspected to be contaminated as they may represent a public health hazard. Biotoxicologic tests in use are laboratory animals (mouse tests) and quadruplex PCR to find the spore-producing toxin types A, B, E, and F. It is also important to follow the patient after clinical recovery to evaluate for persistent toxin and spores in biological samples. 26 Because botulism represents a serious life-threatening condition, patients are often transferred to tertiary care hospitals, adding steps that may further complicate the collection of samples and delay the final diagnosis.
Treatment consists of intensive care support including mechanical ventilation and gavage feeding if needed. Complications are mostly related to mechanical ventilation (pneumonia, atelectasis, tracheitis, granuloma, and tracheal stenosis) and to prolonged hospitalization (nosocomial infections, pressure sores, and deep vein thrombosis).
In the United States, equine and human antitoxin immunoglobulins (botulism immune globulin intravenous human [BIG-IV]) are available and mainly used for infant botulism. In countries where BIG-Ig is not available, plasmapheresis should be considered as a possible alternative to remove the spore or the toxin more rapidly from the circulation. 27
In conclusion, sudden descending symmetrical cranial nerve palsies with generalized weakness or hypotonia should raise suspicion for botulism in all ages of patients. In the absence of a history of ingesting contaminated food or a typical wound, intestinal colonization is a very rare but also underrecognized form of botulism that should be considered both in infants and in older patients.
Footnotes
Acknowledgments
We are indebted to Dr G.M. Svaluto of Pediatric Unit, Hospital Santa Maria della Misericordia, Rovigo, Italy, for clinical assistance to the patient and the Istituto Superiore di Sanità for helping us in performing mouse lethality test and all biotoxicologic tests.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors received no financial support for the research, authorship, and/or publication of this article.