
Abstract
Acute hypokalemic paralysis is characterized by muscle weakness or paralysis secondary to low serum potassium levels. Neurogenic diabetes insipidus (DI) is a condition where the patient excretes large volume of dilute urine due to low levels of antidiuretic hormone. Here, we describe a patient with neurogenic DI who developed hypokalemic paralysis without a prior history of periodic paralysis. A 30-year-old right-handed Hispanic male was admitted for refractory seizures and acute DI after developing a dental abscess. He had a history of pituitary adenoma resection at the age of 13 with subsequent pan-hypopituitarism and was noncompliant with hormonal supplementation. On hospital day 3, he developed sudden onset of quadriplegia with motor strength of 0 of 5 in the upper extremities bilaterally and 1 of 5 in both lower extremities with absent deep tendon reflexes. His routine laboratory studies revealed severe hypokalemia of 1.6 mEq/dL. Nerve Conduction Study (NCS) revealed absent compound motor action potentials (CMAPs) with normal sensory potentials. Electromyography (EMG) did not reveal any abnormal insertional or spontaneous activity. He regained full strength within 36 hours following aggressive correction of the hypokalemia. Repeat NCS showed return of CMAPs in all nerves tested and EMG revealed normal motor units and normal recruitment without myotonic discharges. In patients with central DI with polyuria, hypokalemia can result in sudden paralysis. Hypokalemic paralysis remains an important differential in an acute case of paralysis and early recognition and appropriate management is key.
Case Report
A 30-year-old Hispanic male with a history of pituitary adenoma resection at the age of 13, with subsequent pan-hypopituitarism, epilepsy, and medication noncompliance was admitted with seizures and acute diabetes insipidus (DI). During the hospitalization, he remained seizure free on levetiracetam 1 g q12H and was treated with appropriate antibiotics for a dental abscess. He was initiated on a stress dose steroid taper, restarted on hormone replacement and desmopressin replacement therapy for DI.
The patient’s initial hospital stay was without complication until hospital day 3 when a dose of desmopressin acetate (DDAVP) was delayed, and he developed acute quadriplegia. Magnetic resonance imaging (MRI) brain and computed tomography (CT) brain did not reveal any acute abnormalities. Routine laboratory studies were notable for acute hypokalemia from 3.8 to 1.6 mEq/dL (Table 1), which he developed over 12 hours. His thyroid studies were suggestive of secondary hypothyroidism with low thyroid-stimulating hormone level, free T3, total T3 level, and normal free T4 level (Table 1).
Summary of Laboratory Data.a
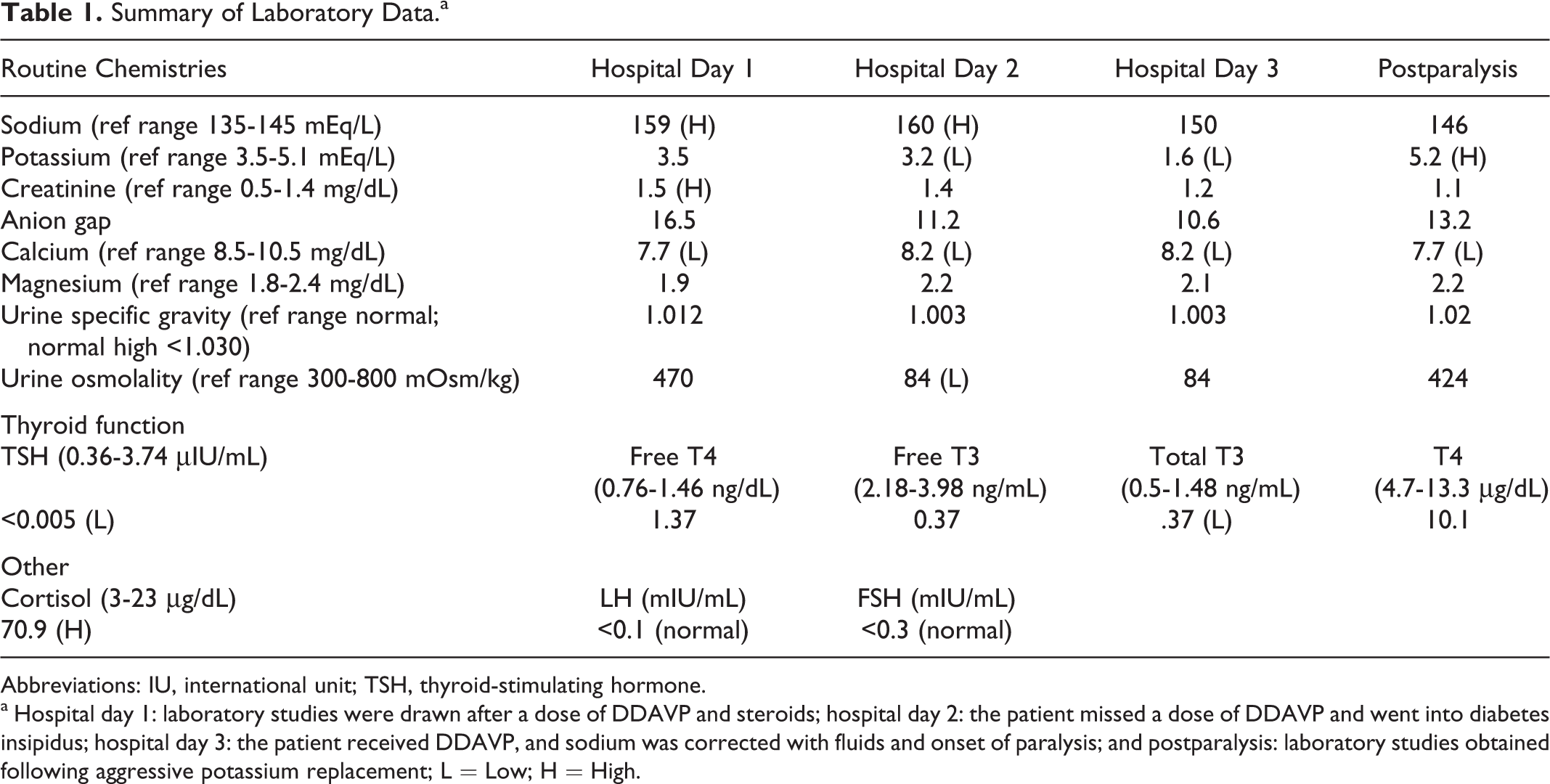
Abbreviations: IU, international unit; TSH, thyroid-stimulating hormone.
a Hospital day 1: laboratory studies were drawn after a dose of DDAVP and steroids; hospital day 2: the patient missed a dose of DDAVP and went into diabetes insipidus; hospital day 3: the patient received DDAVP, and sodium was corrected with fluids and onset of paralysis; and postparalysis: laboratory studies obtained following aggressive potassium replacement; L = Low; H = High.
On examination, he was afebrile and in no apparent distress. He was fully oriented with intact cranial nerves. Muscle strength was 0 of 5 in the bilateral upper extremities, 0 of 5 in the proximal lower extremities, and 1 of 5 in the distal lower extremities. Sensory evaluation of large and small fiber nerves was intact and there was no spinal sensory level. Muscle stretch reflexes were absent in bilateral biceps and triceps and 1+ at bilateral patellae and ankles. No percussion myotonia or cramps were noted. Initial electrodiagnostic studies were performed within 24 hours of paralysis and revealed normal sensory potentials with absent compound motor action potentials (CMAPs; Table 2). Concentric needle electromyography (EMG) examination revealed normal insertional activity with no volitional motor units in the muscles tested and decreased recruitment within distal small motor units. No myotonic discharges or abnormal spontaneous activity were noted.
Summary of Nerve Conduction Studies During Paralysis and Postparalysis.
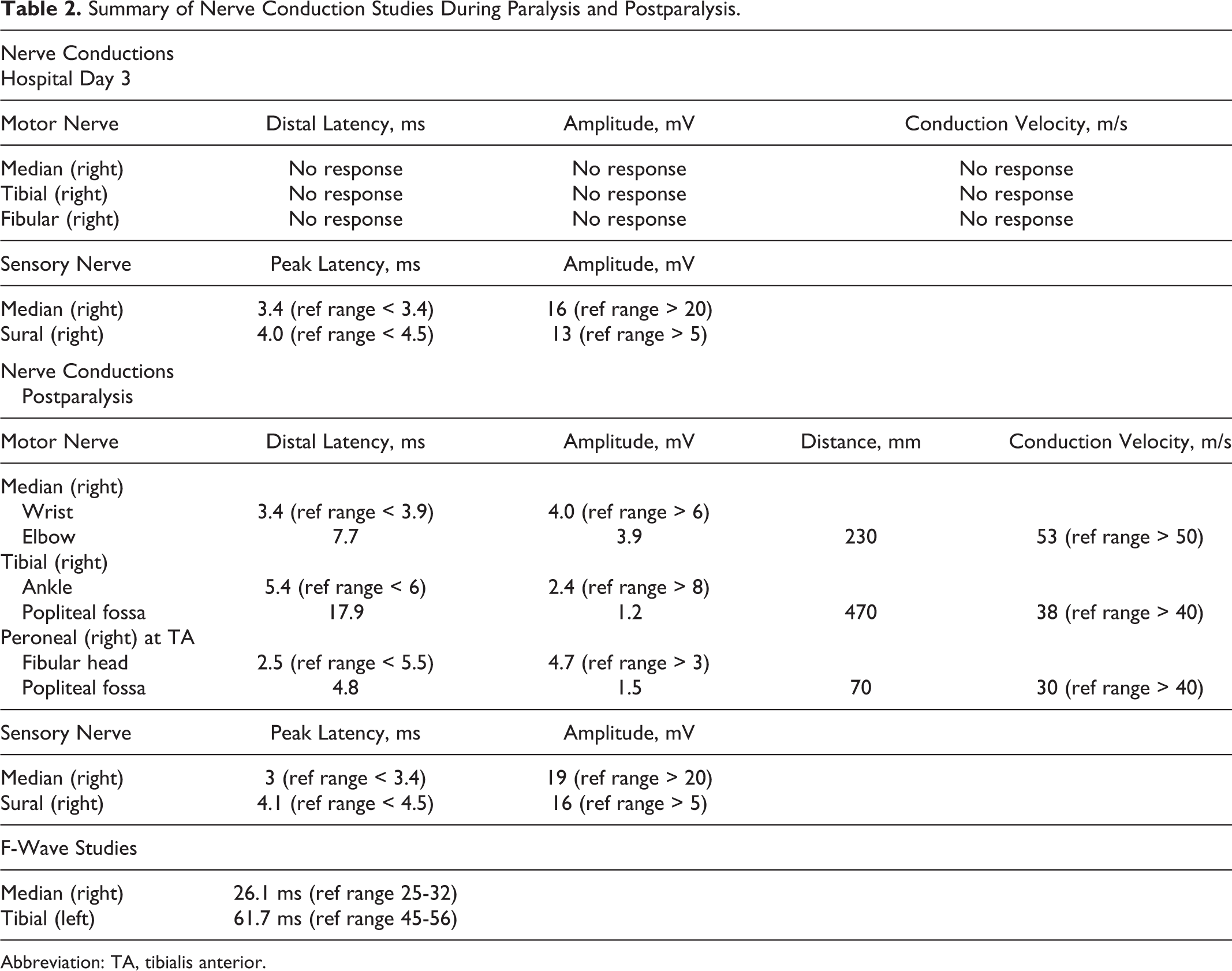
Abbreviation: TA, tibialis anterior.
Hypokalemia was aggressively corrected from 1.6 to 5.2 mEq/dL over 24 hours (potassium chloride 60 mEq over the first 8 hours and 160 mEq within the following 24 hours and potassium phosphate 18 mmol/L within first 12 hours and 36 mmol/L during the second 24 hours; Table 1). Repeat physical examination revealed 5 of 5 strength in all 4 extremities, with normal reflexes. He was later able to ambulate and participate with physical therapy. Repeat electrodiagnostic studies were performed within 48 hours of the initial paralysis. Nerve conduction studies showed return of CMAPs in all limbs, although there was evidence of a demyelinating and axonal polyneuropathy involving the motor more than the sensory nerves (Table 2). Needle EMG did not show abnormal spontaneous activity or myotonic discharges. Previously tested muscles now had normal recruitment and no motor unit abnormalities. Given the findings of a polyneuropathy, it was recommended for him to have further neurological workup as an outpatient, but the patient was lost to follow-up.
Case Discussion
We are reporting a case of paralysis due to hypokalemia in the setting of acute neurogenic DI. Our patient had chronic postsurgical pan-hypopituitarism and was erratic with his medication use. He presented to the hospital with seizures, hypovolemic shock, and metabolic acidosis and was treated with his home dose of DDAVP, fluids and stress dose steroids prior to the collection of his initial laboratory workup. Over the next 2 days, he was treated with DDAVP and fluids with return to normal physiologic parameters (Table 1). Unfortunately, 1 dose of DDAVP was delayed, and the patient relapsed into DI with serum sodium increasing to 160 mEq/dL in conjunction with a low urine osmolality and specific gravity. During this time the patient was polyuric with a urine output of greater than 1 L/h for at least 1 hour. The DDAVP was immediately administered, and aggressive hypotonic fluid resuscitation was resumed. Over the next 12 hours, a positive fluid balance was achieved with appropriate sodium correction to 150 mEq/dL. Since this had been an acute change in sodium level from baseline, rapid correction was deemed safe in this circumstance (Table 1). Unfortunately, the patient developed quadriplegia in this interval. The initial concern was that rapid correction of the sodium had resulted in central pontine myelinolysis, but this was ruled out on emergent MRI. Serum potassium levels were noted to have precipitously fallen from 3.8 mEq/dL prior to DDAVP to 1.6 mEq/dL over a span of 12 hours. In the absence of any other cause of hypokalemia (ie, gastrointestinal losses, insulin infusion), we believe that the etiology was acute renal loss in the absence of any antidiuretic hormone effect (Table 1). Given the reduction in the total body potassium during diuresis, the subsequent resuscitation diluted the serum potassium resulting in the acute systemic hypokalemia manifesting as neuronal dysfunction. This was confirmed by the return of muscle strength, tendon reflexes, and CMAPs upon potassium repletion.
The diagnosis of hypokalemic paralysis is based on a combination of history, physical examination, laboratory and ancillary studies. Patients typically present with acute onset of paralysis with positive family history in most cases. 1 Laboratory studies should be consistent with low serum potassium levels. Acute vascular involvement should be ruled out based on neuroimaging with CT or MRI. The weakness in hypokalemic paralysis can range from mild isolated transient involvement of specific muscle groups to diffuse generalized weakness. Acute cases can last from several hours to days and can be a onetime event or recurrent periodic episodes. 2 Although the majority of cases of acute hypokalemic paralysis are familial (eg, hypokalemic periodic paralysis [HypoPP]), sporadic cases secondary to endocrine, renal, metabolic, and iatrogenic etiologies are often seen (Table 3). 3,4 Studies examining the utility of potassium–creatinine (K/C) ratio and transtubular potassium concentration gradient (TTKG) in further characterizing hypokalemic paralysis have concluded that K/C values above 2.5 and TTKG above 3.0 are suggestive of secondary HypoPP. 1
Secondary Causes of Hypokalemic Paralysis.

Among the familial hypokalemic paralyses, HypoPP is a rare disorder classified as a channelopathy with weakness or paralysis associated with low serum potassium levels. 1 The differential of channelopathies includes several entities and accurate diagnosis can often be challenging, 5 since the clinical presentation varies based on the evaluation setting (ie, outpatient or inpatient). On laboratory evaluation, patients have hypokalemia (which should be normal between attacks) without evidence of thyroid dysfunction (ie, hyperthyroidism) or cardiac dysrhythmia. 1,2 Aggravating or precipitating factors include infection, metabolic, hormonal (ie, menstruation), stress (ie, sleep disturbance), and medications (eg, β-agonists, insulin, corticosteroids). 2 HypoPP can be grouped into paralytic and myopathic cases. 2 Respiratory and cardiac muscles are usually spared in the paralytic form. The myopathic phenotype is seen in 25% of the cases and is characterized by exercise intolerance and progressive fixed muscle weakness in the lower extremities. 2
Thyrotoxic periodic paralysis is another differential when evaluating hypokalemic paralysis. 1,2,3 There are various theories regarding the mechanism of the hypokalemia in an elevated thyroid state, including the direct effect of high circulating hormone titers, but the exact mechanism remains unclear. 1 In these cases, the low potassium needs to be corrected along with antithyroid medications. Our patient did not have elevated thyroid enzyme levels, and his thyroid panel was suggestive of secondary hypothyroidism.
Potassium supplementation is warranted in acute cases of hypokalemic paralysis. Oral replacement with potassium chloride is usually preferred over intravenous supplementation. Intravenous potassium should be used in cases with cardiac arrhythmia or respiratory compromise secondary to pharyngeal weakness or involvement of respiratory muscles. 1 A recommended initial dose of 0.5 to 1 mEq/kg would raise serum potassium concentrations by 1 to 1.5 mEq/L. 1 Prophylactic management with carbonic anhydrase inhibitors (CAIs) such as acetazolamide can be considered in cases with frequent, recurrent attacks. 6,7 In patients with limited response to CAIs, potassium-sparing diuretics such as triamterene and spironolactone can be considered. 1 The efficacy of topiramate, an anticonvulsant with CAI-like activity, has been described in a case report. 7
This case illustrates the diagnostic challenge of recognizing an acute neurologic effect of a systemic, metabolic pathology. Our patient had a known diagnosis of neurogenic DI, and his acute hypokalemia was likely a result of polyuria. We acknowledge that this case is limited in that many of the urine and serum studies were drawn after medical intervention, making it difficult to obtain accurate supportive data. In addition, as he did not follow-up for genetic testing or further electrodiagnostic studies, we are unaware whether he had additional episodes of muscle weakness. Hypokalemic paralysis should be considered as a differential in patients presenting with acute flaccid paralysis in the setting of an acute illness. In the hospital setting, and particularly in neurological units, emergent management should include adequate timely potassium replacement, diagnostic evaluation, and close monitoring. 1,6,8
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.