
Abstract
This report explores the case of a 49-year-old African American male with a six-month history of multifocal neurological deficits who presented to an outside hospital after a generalized seizure. Patient was transferred to our tertiary medical center after brain imaging showed multiple bilateral supratentorial intraparenchymal hemorrhages (IPH). A brain biopsy confirmed parenchymal and perivascular non-caseating granulomas with vasculitis. The patient was definitively diagnosed with neurosarcoidosis (NS) and his condition improved with high dose corticosteroids and additional immunosuppressive therapies. Intracranial hemorrhage in the setting of NS is extremely rare, with fewer than thirty documented cases; however, this is likely an underestimation of its true prevalence. This case illustrates the difficulty in diagnosis as many other etiologies of IPH must be considered. Additionally, the clinical course and manifestations of NS is often quite variable. The uniqueness of this case lies in the rapid progression from seemingly incidental microhemorrhages to multiple large IPHs over two months. While the cause of this progression is not immediately apparent, a possible cause may be inadequate initial treatment due to delayed diagnosis. Our case demonstrates the importance of early recognition and initiation of immunosuppressive therapy, potentially leading to dramatic clinical improvement, as seen in this patient.
Keywords
Background
Neurosarcoidosis is a rare central nervous system (CNS) complication reported in 5% of systemic sarcoidosis cases, with intracranial hemorrhage presenting in as few as 1% of all NS patients.1-3 Because of its infrequency, intracranial hemorrhage in NS presents as a formidable diagnostic challenge with poorly defined management criteria. We report the case of a gentleman with a six-month history of multiple neurological complaints who initially presented to an outside hospital after a generalized seizure. The patient was transferred to our tertiary medical center after brain imaging showed multiple bilateral supratentorial intraparenchymal hemorrhages. Subsequent brain biopsy showed nonnecrotizing perivascular and parenchymal granulomas.
Case Report
The patient is a 49-year-old African American man with a past medical history of hypertension and diabetes, who presented to an outpatient neurologist for six months of bilateral lower extremity weakness, left sided hearing loss, urinary urge incontinence, and gait instability. Prior to his Neurology visit, the patient had a lumbar spine MRI without contrast which showed abnormal T2 signal at distal thoracic cord/conus. Subsequent MRI brain showed mild periventricular subcortical white matter hyperintensity and punctate bilateral supratentorial hemosiderin deposition. MRI lumbar spine with contrast showed abnormal T1 enhancement at the distal thoracic spine and conus (Figure 1A). Cervical spine imaging was unremarkable. Immunological workup was only significant for elevated Antiproteinase 3 (PR-3) antibodies at 4.8 U/mL (0.0 – 3.5). Intravenous steroids were not initiated at that time because of a history of diabetes and chronicity of symptoms. Instead, the patient was recommended to follow up with a neuroimmunologist at an academic tertiary medical center for further diagnostics.
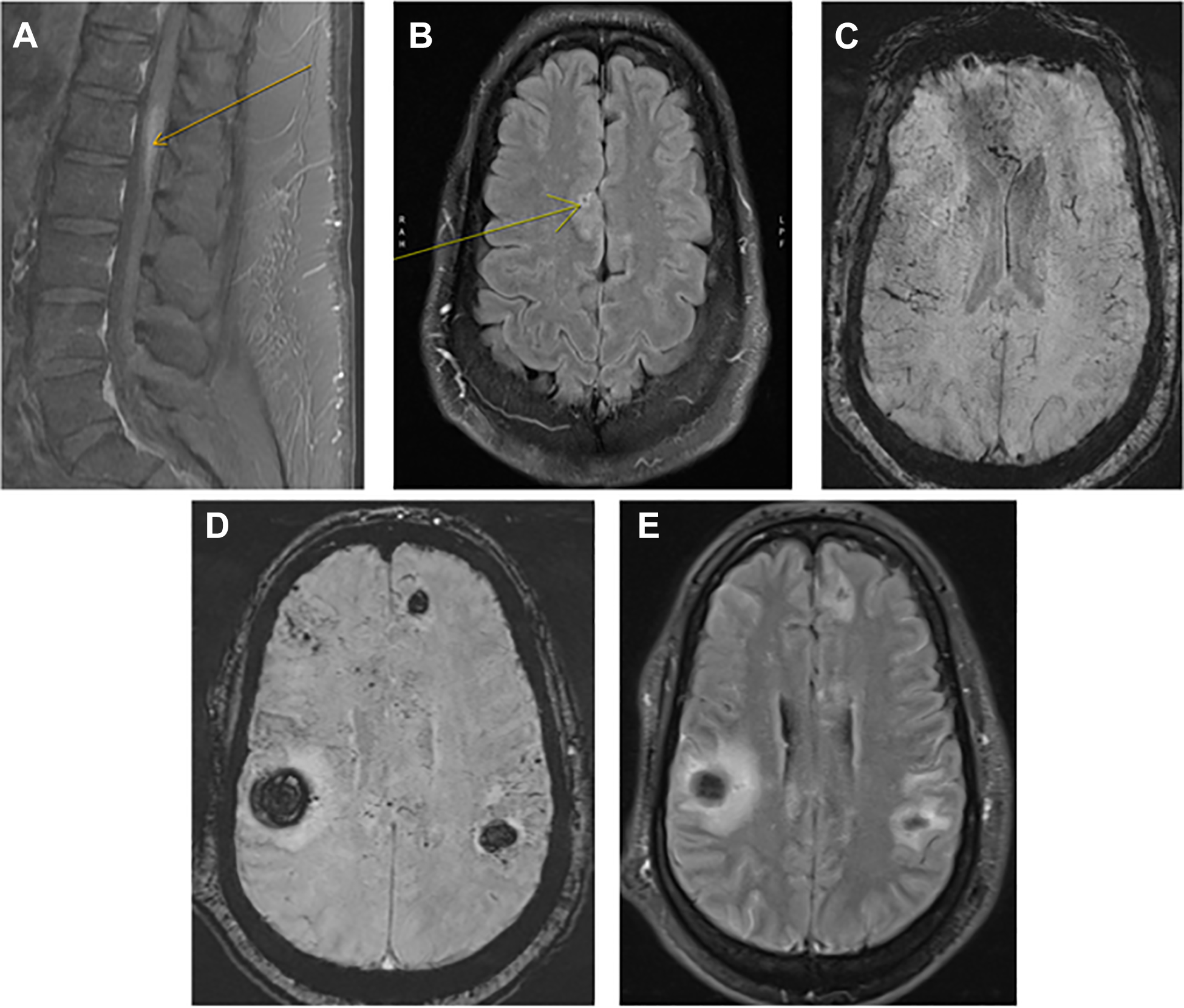
A MRI T1 post contrast of the sagittal lumbar spine showing distal thoracic/conus L1 level expansion and enhancement (see arrow). figure 1B: MRI brain T2 fluid attenuation inversion recovery (FLAIR) post contrast showing minimal leptomeningeal enhancement of the parasagittal right frontal lobe (see arrow). figure 1C: susceptibility weight imaging (SWI) showing extensive microhemorrhages throughout the supratentorial brain and dilated or thrombosed medullary veins. figure 1D: SWI showing multiple scattered intraparenchymal hematomas throughout the supratentorium, and extensive microhemorrhages again appreciated. figure 1E: FLAIR post contrast showing diffuse leptomeningeal enhancement throughout the supratentorial brain.
At his first neuroimmunology office visit one month later, an immediate family history of sarcoidosis and lupus was noted. Repeat MRI brain with contrast showed extensive punctate areas of enhancement throughout the supratentorium, extensive chronic microhemorrhages, and minimal leptomeningeal enhancement of the parasagittal right frontal lobe (Figures 1B and 1C). CT chest with contrast showed small bilateral paratracheal, subcarinal, mediastinal, and hilar lymphadenopathy. A complete blood count, comprehensive metabolic panel, inflammatory, and infectious laboratory workup were all unremarkable. Lumbar puncture was discussed but never completed. Steroids were held pending lymph node biopsy by the pulmonologist. Transbronchial lymph node fine needle aspiration was done one week later which showed giant cells and rare aggregates of epithelioid histiocytes suggestive of granulomas, flow cytometry was unremarkable. Patient followed up in the neuroimmunology clinic one month later. Due to the suspicion for vasculitis or sarcoidosis, the patient was started on 60 mg daily of prednisone taper and bridged to methotrexate starting at 2.5 mg weekly. Repeat MRI brain and MRI lumbar spine with contrast was recommended at three months follow up.
Two weeks after his clinic visit, the patient was brought into the emergency room of an outside hospital combative and confused after experiencing a witnessed generalized convulsive seizure while at work. He was given 2 mg IV lorazepam, loaded with levetiracetam 1,500 mg, and placed in physical restraints. Pertinent laboratory data showed hyperglycemia to 164 mg/dL and lactate of 7.4 mmol/L, while troponin, ethanol, acetaminophen, and salicylates were all negative. Head CT showed multiple (six) foci of acute intraparenchymal hemorrhages involving bilateral frontal and parietal lobes, the largest measuring 2.1 x 3.2 cm in the right parietal lobe and associated with mild effacement of the adjacent lateral ventricle.
Patient was intubated and transferred to our neurological ICU. Initial vital signs were HR 100 bpm, BP 155/92 mmHg, RR 18 BR/min, and SaO2 99% on a ventilator. When examined off sedation, patient was awake with eyes open, regarded the examiner, pupils were symmetric and briskly reactive to light with a positive vestibular ocular reflex, strong corneal reflex, strong, and withdrew/grimaced to noxious stimuli in all extremities. The patient was started on a nicardipine drip, levetiracetam 1,000 mg twice daily, and managed conservatively. A complete blood count, comprehensive metabolic panel, serum immunological, and infectious workup was completed and all unremarkable. Repeat PR-3 antibodies were within normal limits. A lumbar puncture performed on day two of hospitalization revealed lymphocytic pleocytosis and CSF cytology showed no evidence of malignancy. A few days into the hospital course, the patient’s neurological exam was unchanged, he remained intubated and sedated. Echocardiogram, CTA head and neck, CT chest/abdomen/pelvis with contrast, diagnostic cerebral angiogram, and EEG were all unremarkable. MRI brain with contrast showed known IPHs, scattered microhemorrhages, and diffuse leptomeningeal enhancement in the supratentorial brain (Figures 1D and 1E). MRI lumbar spine with contrast was unchanged from prior. Based on the patient’s clinical history and neuroimaging, he was started on high dose methylprednisolone 1 g for five days and a right frontal lobe brain biopsy was then performed. After starting steroids, the patient’s neurological exam gradually improved and at the time of discharge to an acute rehabilitation facility he was appropriately conversing with hospital staff and ambulating with a walker. He still experienced urinary incontinence and left sided hearing loss which were both treated with supportive care. Histopathological examination of the brain biopsy pathology revealed scattered parenchymal and perivascular non-necrotizing granulomas and focal non-necrotizing granulomatous vasculitis involving small parenchymal blood vessels and leptomeningeal vessels. There was no fibrinoid necrosis and infectious organisms were not identified (Figure 2 A-C). Based on these findings, a diagnosis of neurosarcoidosis was established. The patient was started on an infliximab infusion regimen of 5 mg/kg, methotrexate, and a high dose prednisone taper. During a follow up appointment two months after discharge, the patient was ambulating without any assistance, had full strength in his lower extremities, no bowel or bladder incontinence, and unchanged left sided hearing loss. A repeat MRI brain with contrast three months after discharge showed resolving IPHs and no leptomeningeal enhancement. Patient was continued on methotrexate, low dose prednisone taper and infliximab infusions every six weeks.
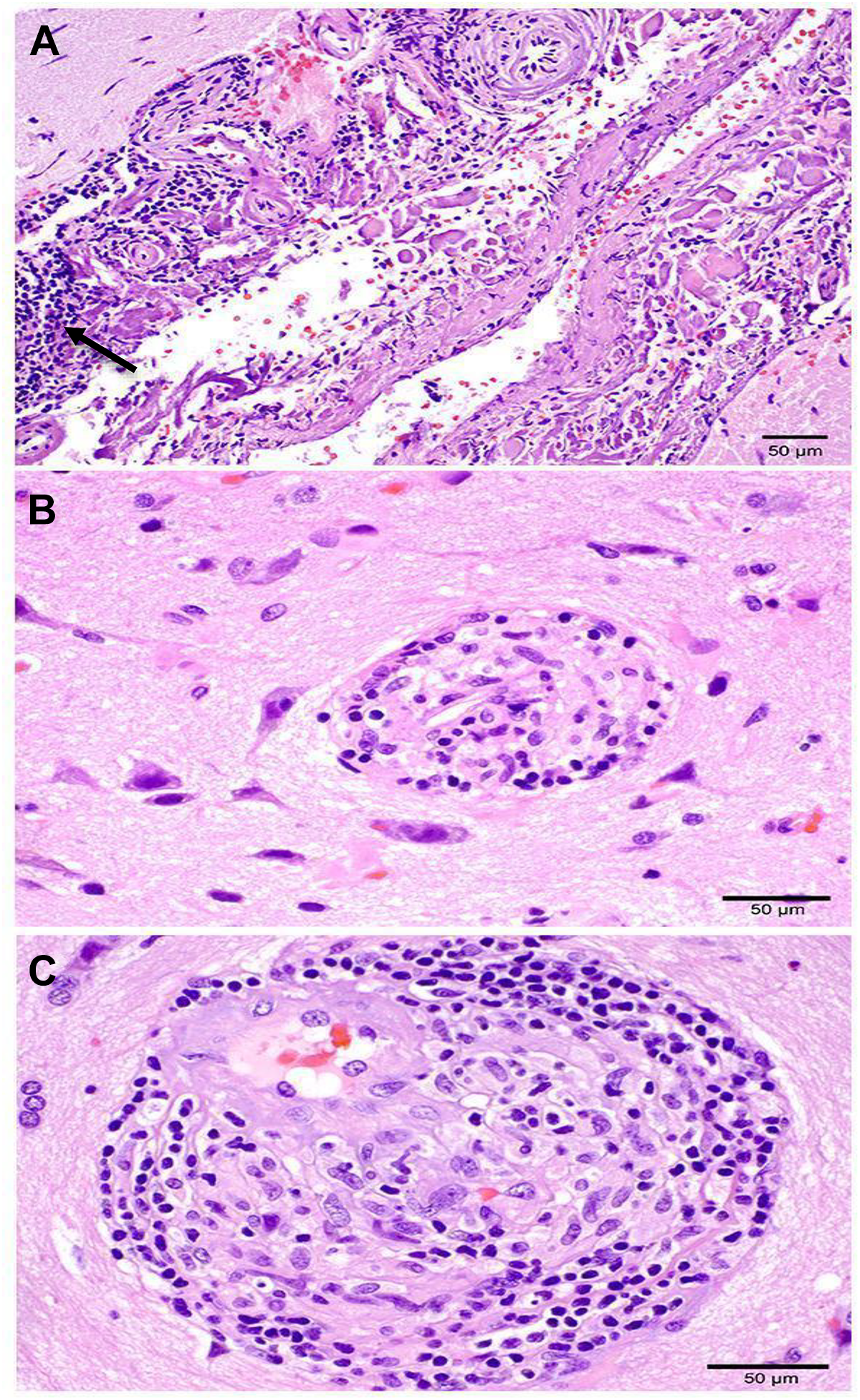
Histologic features of brain biopsy leptomeninges demonstrate dense collagen fibrosis and robust lymphocytic and histiocytic inflammatory infiltrates (see arrow) with focal transmural involvement of large leptomeningeal vessels (A). rare multinucleated giant cells were identified (not pictured). well defined non-necrotizing granulomas composed of lymphocytes and histiocytes were seen in parenchymal (B) and perivascular (C) locations.
Discussion
Intracranial hemorrhage in NS is an exceedingly rare presentation with a reported incidence ranging from <1% to 6% in retrospective reviews.4,5 In our search of the literature, we identified fewer than thirty total reported cases. Cerebrovascular manifestations of NS, including both ischemic and hemorrhagic lesions, are broadly thought to be due to vasculopathy related to the underlying systemic inflammatory disease.4,5 The precise mechanism is not clear, but histopathologic evaluation of hemorrhagic cases has demonstrated both arterial and venous vasculitic involvement by epithelioid cell granulomas and other inflammatory infiltrates.3,5 The ensuing mixed vascular damage may result in either subarachnoid or intraparenchymal hemorrhages. Here, we presented the case of a patient with multiple IPHs along with multifocal microhemorrhages following the development of neurological symptoms of as-yet-unconfirmed NS.
In our patient, the principal differential considerations beyond NS were other causes of IPHs: hypertensive hemorrhage, cerebral amyloid angiopathy, another CNS vasculitis, or systemic vasculitis with secondary CNS involvement. In primary CNS vasculitis, abnormal angiographic findings have been reported in the majority of cases. 3 Our patient had no such abnormalities which is expected in NS because of its microvascular histopathology. 5 Our patient also had no microscopically observed amyloid deposition to suggest amyloid angiopathy as an etiology. Although the patient had a known history of hypertension, the presentation of multiple similarly aged hemorrhages suggested an alternative diagnosis. Tissue biopsy, considered the gold standard for diagnosis of NS, showed non-necrotizing histiocytic granulomas in parenchymal and perivascular locations with rare vessels showing granulomatous vasculitis. MRI imaging also demonstrated leptomeningeal involvement which supported the case for NS. Combined with the characteristic symptom of cranial neuropathies, the chronic, non-localizing neurological symptoms suggested a more systemic neurological disorder. With histopathologic correlation and the reasonable exclusion of alternatives, our diagnosis was established and met a “definite” NS classification per the Zajicek criteria. 6
We reviewed available case reports of intracranial hemorrhage in NS. Our case appears to be unique in the rapid evolution of IPHs that progressed from seemingly incidental microhemorrhages to multiple larger IPHs over a period of two months. Treatment of NS is likely limited by the difficulty and delays in its diagnosis. This is of special importance when dealing with a life-threatening complication such as IPHs. Retrospective data has shown a 33% mortality rate among NS patients with intracranial hemorrhage, compared to reports of 5-10% mortality for remaining NS patients. 5 One such contribution to this mortality would be further complications from the hemorrhage itself, as we saw with our patient’s new onset seizure. Another consideration is the possibility that NS hemorrhages may be underreported in the literature. There are several reasons we suspect this. First, it may be underrecognized due to the uncommon association. Second, it may be the only manifestation of NS at the time of presentation, obscuring the link. Finally, the occurrence of asymptomatic microhemorrhages may lead to underestimation of true prevalence.
There is currently a lack of controlled trials evaluating immunosuppressive therapies in the overall treatment of neurosarcoidosis, much less its unusual variants. To address this shortcoming, we referred to literature focused on the treatment of severe and refractory cases of NS. In such cases, there has been evidence of favorable clinical response following the use of the TNF-α antagonist, infliximab. 7 Previously documented case reports of intracranial hemorrhage in NS had established improvement with our initial strategy of high dose corticosteroids.8,9 Due to our patient’s severe presentation, we elected to pursue a more aggressive, early therapy with infliximab in addition to methotrexate and a steroid taper. We hoped to avoid further complications from disease progression in a patient with an already precarious neurological state. This approach appears to have been successful as demonstrated on follow up examination and imaging. Further studies are needed to fully assess the safety and efficacy of such a strategy, though our case is supportive.
Our case highlights an unusual presentation of neurosarcoidosis where multiple simultaneous intraparenchymal hemorrhages and microhemorrhages were seen following chronic neurological symptoms. Although the overall incidence of NS related intracranial hemorrhage complications appears to be low, it has significant life-threatening potential and is likely underrecognized. Since timely diagnosis has implications on management of the condition, we believe our case may be helpful in characterizing this uncommon disease. Additionally, we have shared our experience with a treatment strategy which was able to achieve both clinical and radiologic improvement.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.