
Abstract
This article characterizes 2 cases of longitudinally extensive transverse myelitis (LETM) that did not respond to immunotherapy and were diagnosed by biopsy as primary central nervous system (CNS) malignancies. Diffuse H3 K27M-mutant glioma is a recently described entity with very few cases of isolated spinal disease described in adults. Primitive neuroectodermal tumor is similarly uncommon in the spinal cord. Malignancies should be considered in patients who fail to improve with immunomodulatory therapy. We believe the experiences of our center will raise awareness about that point, broaden the existing understanding of the diagnostic approach to LETM, and highlight the need for additional studies.
Keywords
Introduction
Longitudinally extensive transverse myelitis (LETM) is defined as myelitis spanning 3 or more vertebral bodies. In young adults, acute to subacute isolated LETM raises suspicion for anti-aquaporin-4 IgG (AQP4-IgG) seropositive neuromyelitis optica spectrum disorder (NMOSD) or myelin oligodendrocyte glycoprotein (MOG)-IgG related disease. In both disorders, patients typically reach a clinical and radiographic nadir within weeks of onset, and persistent enhancement raises suspicion for alternative diagnoses, including neurosarcoidosis and spinal cord malignancy.
Spinal cord tumors represent 2-4% of primary central nervous system (CNS) tumors, with metastatic tumors vastly more common than primary spinal tumors in adults. 1 Here, we present 2 unusual cases of primary spinal cord tumors presenting with acute LETM in young adult patients.
Case 1
A previously healthy 21-year-old woman presented with 1 week of right leg weakness and urinary incontinence. Initial neurological examination was notable for hypoesthesia and weakness of the right lower extremity. MRI of the thoracic spine showed a T2 hyperintense intramedullary lesion extending from T9-T12 with marked edema and peripheral gadolinium enhancement (Figure 1A-C) suggestive of LETM. No brain or cervical spinal lesions were present. Cerebrospinal fluid (CSF) studies were unrevealing, with protein 30 mg/100 mL (normal 15-60 mg/100 ml), glucose of 61 mg/100 mL (normal 50-80 mg/100 ml), and 1 total nucleated cell (normal 0-5 cells). IgG index was not elevated, no oligoclonal bands were identified, and no abnormal cells were appreciated on cytology. Serum anti-AQP4 and anti-MOG antibodies were negative. She was treated with methylprednisolone 1g/day for 5 days followed by 5 sessions of plasmapheresis with no improvement. Spine MRI 3 months later showed increased edema and persistent contrast enhancement (Figure 1D-E) and the neurological exam was unchanged. Full-body positron emission tomography (PET) showed hypermetabolic uptake in the thoracic spine lesion but no other abnormalities. Biopsy showed a grade IV H3 K27M-mutant (IDH negative, MGMT unmethylated) glioma. She received spinal radiation concurrent with Panobinostat.
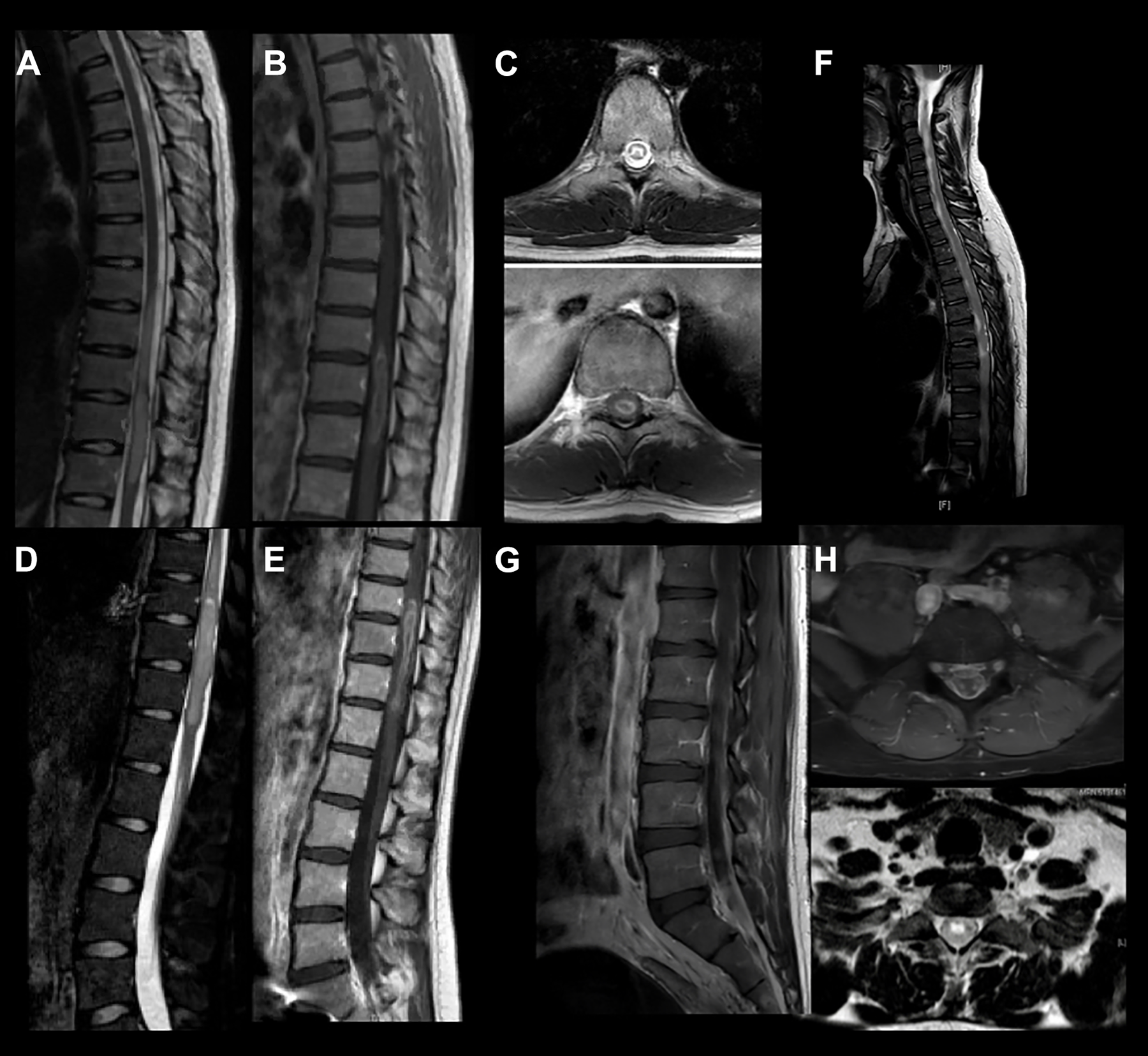
Case 1: MRI of the thoracic spine showed (A) a T2 hyperintense intramedullary lesion extending from T9-T12 with marked edema and (B-C) peripheral gadolinium enhancement. (D-E) Repeat MRI 3 months later demonstrated increased edema and persistent enhancement. Case 2: Spine MRI revealed (F) an expansile intramedullary lesion extending from the cervicomedullary junction to the conus with (G-H) additional intramedullary lesions involving the cauda equina.
Case 2
A previously healthy 31-year-old man presented with 3 weeks of progressive left leg pain and paresthesias as well as urinary retention. Initial neurological examination was notable for left lower extremity weakness, diffuse hyperreflexia, and a positive left Babinski sign. Spine MRI demonstrated an expansile intramedullary lesion extending from the cervicomedullary junction to the conus, with additional intramedullary lesions involving the cauda equina (Figure 1F-H). Full-body PET revealed hypermetabolic uptake within the spinal cord lesions but no other abnormalities. Scrotal ultrasound was unrevealing. CSF studies were notable for elevated protein of 392 mg/dL (normal 15-60 mg/100 ml) and decreased glucose of 13 mg/dL (normal 50-80 mg/100 ml). Flow cytometry was unremarkable, but cytology demonstrated atypical cells. He underwent lumbosacral laminectomy with biopsy, which revealed malignant round cells with neuro-endocrine differentiation, with final pathology consistent with CNS embryonal tumor. He received craniospinal radiation concurrent with carboplatin and vincristine chemotherapy.
Methods
In accordance with Boston Medical Center policy regarding case series involving 3 or fewer patients, Institutional Review Board approval was waived. Cases were reviewed for HIPAA compliance and privacy.
Discussion
We describe 2 young patients whose presentations were initially suspicious for NMOSD and or another inflammatory etiologies of LETM before they were diagnosed with primary high-grade malignancies of the spinal cord.
Primary high-grade malignancy of the spinal cord without involvement of the brain or body is vanishingly rare (3.1% of the primary CNS tumors in the US). In adults, ependymomas are the most common intramedullary spinal cord tumors, followed by astrocytomas and oligodendrogliomas. High-grade astrocytomas account for less than 10% of intramedullary spinal cord tumors in both children or adults. 1 Grade IV diffuse H3 K27M-mutant gliomas occur primarily in children but have been described in patients up to 65 years old. 2 These rare tumors, only incorporated to the World Health Organization (WHO) classification in 2016, tend to occur in the thalamus and pons and carry a poor prognosis. 2 Only 21 cases of diffuse H3 K27M-mutant glioma isolated to the spinal cord have been reported in the literature. 2 -4
CNS embryonal tumors are a heterogeneous group of malignant neoplasms found in both the central and peripheral nervous systems, 5 representing less than 1% of primary spinal cord malignancies. 6 The 82 cases reported to date are primarily of children and young adults, but patients up to 70 years old have been described. 5 Primary spinal CNS embryonal tumors carry a poor prognosis despite maximal therapy, which may include surgical resection alongside adjuvant chemotherapy and radiotherapy. 7
Although rare, primary spinal cord malignancy should remain on the differential in young adults presenting with LETM given the potential for high morbidity and mortality and the opportunity for directed treatment. CSF studies tend to show higher protein levels and lower cell counts in neoplasms compared to inflammatory myelopathies, but there is significant overlap in the ranges of these parameters. 8 In NMOSD flares, about 85% of patients improve with first line immunosuppressive treatment—typically high-dose intravenous steroids for 5 days and/or plasma exchange. 9 Patients who continue to worsen after 21 days or fail to improve with immunomodulatory therapy should have repeat imaging.
Spinal cord biopsy is a risky, uncommon procedure without clear criteria for its indications. The rate of complications is close to 20% and diagnostic yield is low at approximately 25%. 8 It can be avoided in the vast majority of cases with extensive medical workup and empirical therapy. One may consider biopsy in cases where extensive workup is non-diagnostic, there are progressive deficits in spite of immunomodulatory therapy, and radiographic enhancement persists for greater than 3 months.
Footnotes
Authors’ Note
Lucas Horta and Deepti Virmani contributed to design and conceptualization of study; collection and analysis of data; and drafted manuscript for intellectual content. K. H. Vincent Lau and Pria Anand contributed to design and conceptualization of study; analysis of data; revision of manuscript for intellectual content; and study supervision. This work is not currently under review at any other journal and has not previously been published. In accordance with Boston Medical Center policy regarding case series involving three or fewer patients, Institutional Review Board approval was waived. Cases were reviewed for HIPAA compliance and privacy.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.