
Abstract
We report two distinct challenging initial presentations of myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Case 1 describes a 12-year-old boy who developed headaches refractory to pain medication followed by cranial neuropathies and intracranial hypertension, confirmed by lumbar puncture with an opening pressure >36 cm H2O. Case 2 describes a 3-year-old boy who developed new-onset seizures refractory to antiseizure medications, a presentation of FLAIR-hyperintense lesions in MOG-antibody associated encephalitis with seizures (FLAMES). On repeat magnetic resonance imaging, both patients were found to have cortical T2 hyperintensities, leptomeningeal contrast enhancement, and bilateral optic nerve enhancement. In the cerebrospinal fluid, both patients had CSF pleocytosis with neutrophilic predominance. The patients were treated with intravenous immunoglobulins, plasma exchange, and high-dose corticosteroids. The first patient achieved disease remission, whereas the second patient required the addition of rituximab for management of seizures. The two cases highlight the pleomorphic clinical phenotypes of MOGAD.
Keywords
Introduction
The expression of myelin oligodendrocyte glycoprotein (MOG) on the outer lamella of central nervous system myelin sheath makes it an accessible target for autoantibodies. 1 MOG antibody-associated disease (MOGAD) represents a growing constellation of neurological syndromes evocative of multiple sclerosis and neuromyelitis optica spectrum disorder. The latest proposed diagnostic criteria for MOGAD include having a (1) presentation of acute disseminated encephalomyelitis (ADEM), optic neuritis (ON), myelitis, cerebral focal deficit, brainstem syndrome, or cortical encephalitis often with seizures, (2) positive serum MOG-IgG test by cell-based assay, and (3) exclusion of a better diagnosis. 2 If serum MOG-IgG is low-positive, positive without a titer, or only CSF positive, then it still meets criteria (2) as long as serum aquaporin-4-IgG (AQP4-IgG) is negative and the patient has clinical or MRI features of optic neuritis, myelitis, or brain/brainstem syndrome. 2 There remains a growing number of clinical presentations that continue to be reported. Here, we describe two pediatric cases of MOGAD with confounding initial presentations and highlight their diagnostic challenges.
Case 1
A 12-year-old right-handed obese boy presented to the emergency department with 2-week history of worsening right frontal headache and pain throughout the neck and back. His neurologic examination and brain MRI with and without contrast were unremarkable, and he was discharged on naproxen with minimal relief. He returned a week later with worsening headache and drowsiness, rapidly progressive blurry vision, facial droop, slurred speech, and imbalance. His neurologic examination was remarkable for 20/30 visual acuity bilaterally (baseline 20/20), left lower facial weakness, severe dysarthria, left Babinski, and ataxic gait. Fundus examination revealed bilateral disc edema. Brain MRI without contrast revealed multifocal cortical/subcortical T2 hyperintensities, bilateral optic nerve head elevation, and enlarged, T2 hyperintense intraorbital optic nerves (Figure 1A-F). MR angiogram of head and neck and MRI of entire spine were normal. Case 1 neuroimages. (A)-(F) MRI brain without contrast. (A), (B) Axial FLAIR shows bilateral, multifocal T2 hyperintense cortical edema (arrows) in the frontal and temporal lobes. (C) Axial DWI shows no diffusion restriction. (D) Axial T2 of orbits reveals optic nerve head elevation (arrow), left > right. E Axial FLAIR of orbits reveals T2 hyperintensity of the bilateral intraorbital optic nerves (arrows) with F facilitated diffusion on DWI sequence. (G)-(K) Follow-up MRI with and without contrast after 3 weeks. (G) Axial postcontrast FLAIR shows decreased frontotemporal T2 hyperintense cortical edema and (H) newly apparent scattered leptomeningeal enhancement (arrows). (I) Axial T1 postcontrast shows left frontal leptomeningeal enhancement (arrow). (J) Axial postcontrast FLAIR and (K) T1 fat-saturated images of orbits show bilateral intraorbital optic nerve and optic nerve sheath T2 hyperintensity and enhancement (upper arrows), respectively. Prechiasmatic optic nerves and optic chiasm were normal (lower arrows).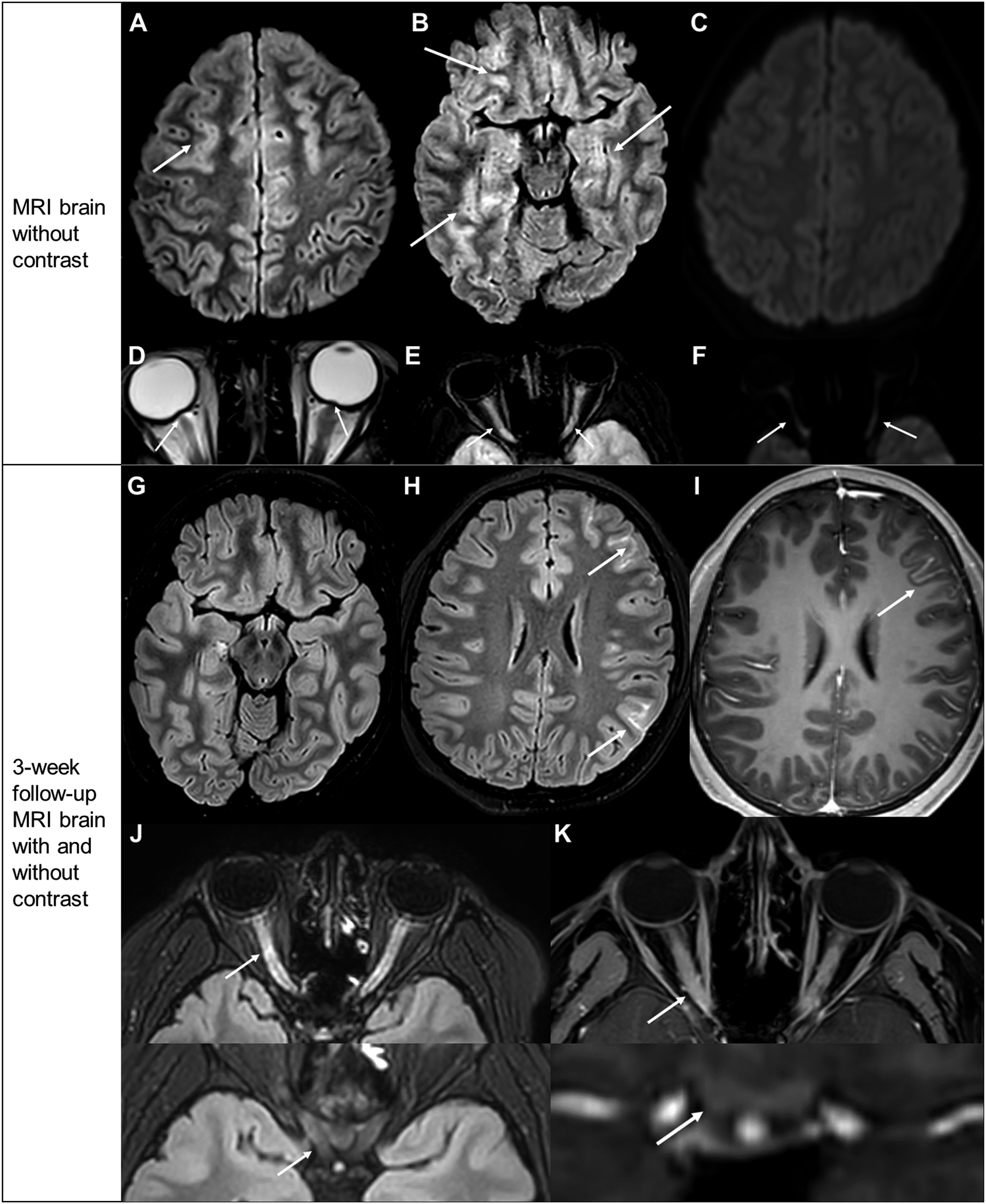
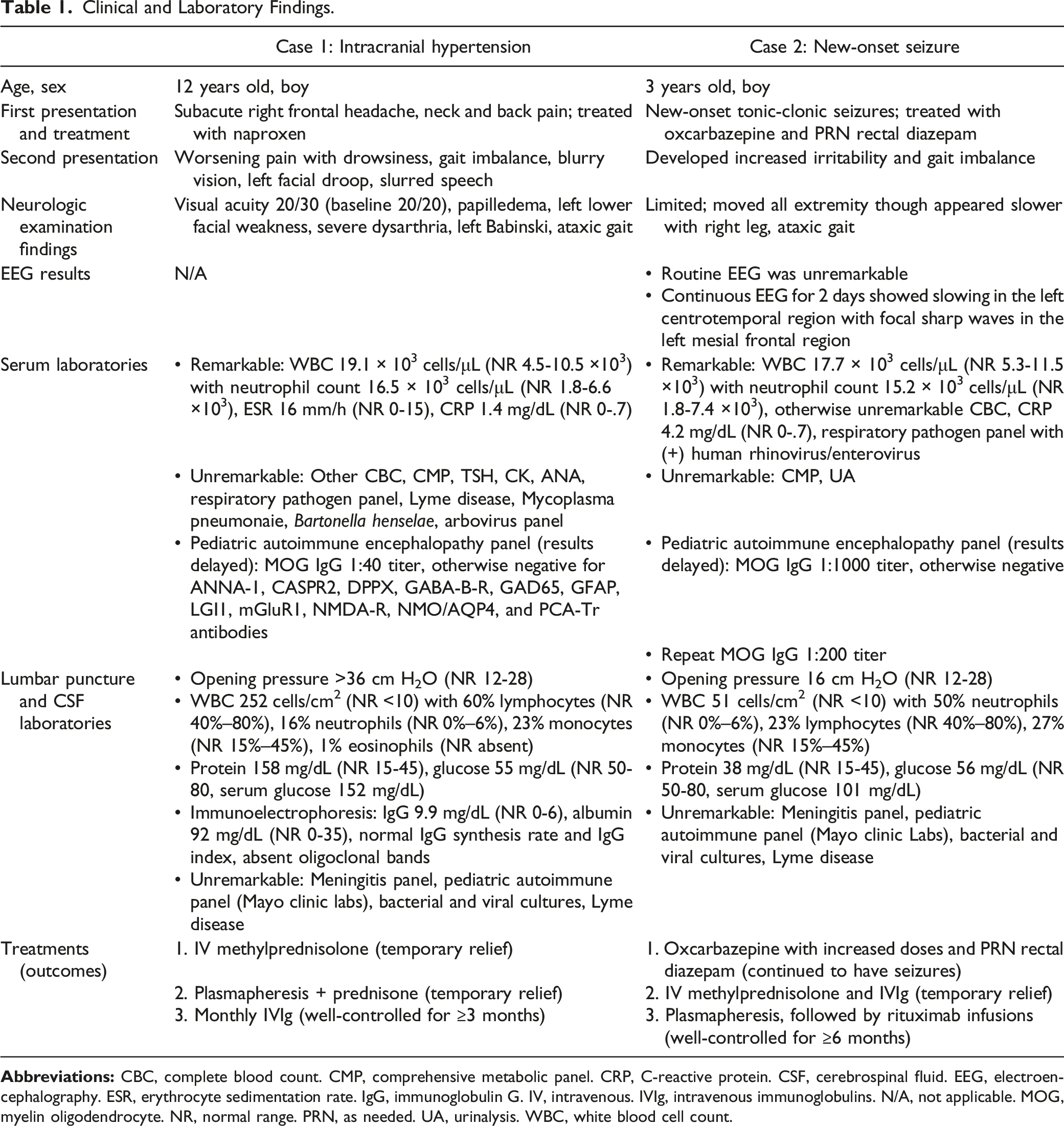
Clinical and Laboratory Findings.
Two weeks later, he returned with ocular motor pain and acute vision loss, described as a “dark curtain rising” through the right eye and blurry vision with the left. Fundus examination revealed mild optic nerve pallor in the right eye. No papilledema was observed. MRI of the brain and orbits with and without contrast showed improvement in the bilateral frontotemporal T2 hyperintense edema, but revealed scattered foci of left frontoparietal leptomeningeal enhancement (Figure 1G-I). The intraorbital optic nerve segments appeared diffusely enlarged, T2 hyperintense, and contrast-enhancing with sparing of the chiasm, consistent with ON (Figure 1J-K). He received plasmapheresis every other day with corticosteroids on days off for two weeks with vision improvement.
The serum pediatric autoimmune encephalopathy panel from before revealed low-positive MOG-IgG (1:40 titer, live cell-based assay) and negative AQP4-IgG. With his accompanying clinical and radiographic findings of optic neuritis and demyelinating cortical lesions, he met diagnostic criteria for MOGAD. 2 He received 5 rounds of plasmapheresis and then was discharged on a long oral corticosteroid taper and monthly intravenous immunoglobulin therapy (IVIg). On 3-month follow-up, he remained in remission, with intact visual acuity and normal fundus examination.
Case 2
A 3-year-old ambidextrous boy presented to the emergency room after a new-onset tonic-clonic seizure at daycare with emesis and left gaze deviation. He had postictal agitation but soon returned to baseline. Initial workup showed a mild leukocytosis and unremarkable head CT. He was discharged with rectal diazepam as needed for breakthrough seizures. Three days later, he became unresponsive, appeared pale, vomited, and stared off blankly. He again returned to baseline but was admitted to the hospital for evaluation. Routine EEG was normal. Brain MRI with and without contrast revealed asymmetric, right predominant leptomeningeal enhancement, with small foci of T2 hyperintensity in the right temporoparietal subcortex (Figure 2A-F). Due to low suspicion of infectious encephalitis, he was discharged with oxcarbazepine and rectal diazepam, with plans to repeat MRI in 2-3 months. Case 2 neuroimages. (A)-(F) MRI brain with and without contrast. (A), (B) Sagittal postcontrast FLAIR MR images show leptomeningeal enhancement along right temporal sulci and scattered in right frontoparietal lobe (arrows). Additional foci of nonenhancing T2 hyperintensity are observed in the subcortical white matter of the right temporal lobe (dashed arrow). (C), (D) Coronal postcontrast FLAIR MR images show nonenhancing patchy T2 hyperintense foci in subcortical white matter (arrow) and leptomeningeal enhancement in inferior temporal lobe (dashed arrow). (E), (F) Axial postcontrast FLAIR MR images show leptomeningeal enhancement along right precentral sulcus (arrow). Small patchy foci of nonenhancing T2 hyperintensity are observed in the subcortical white matter of the right temporal lobe (dashed arrow). (D)-(M) Follow-up MRI brain and spine with and without contrast, 12 days after prior MRI. (D)-(F) Axial postcontrast FLAIR shows new leptomeningeal enhancement in left central sulcus and temporal sulci (solid arrows). Other small, patchy nonenhancing T2 hyperintense foci are present in the right temporal periventricular white matter (dashed arrows). (G)-(I) Coronal postcontrast FLAIR reveals new leptomeningeal enhancement (arrows) along left Sylvian fissure and temporal sulci. There are also new, patchy foci of subcortical and periventricular white matter nonenhancing T2 hyperintensity in right frontal and parietal lobes (dashed arrows). (J) Axial postcontrast FLAIR and (K) postcontrast T1 of optic nerves show T2 hyperintensity and enhancement of the intraorbital optic nerves and nerve sheaths, left greater than right. There is associated (L) facilitated diffusion in the abnormal segments, which was new compared to prior exam. (M) Sagittal T2-STIR of spine shows central, nonenhancing, mildly expansile T2 hyperintensity within the spinal cord from C4 through T3 (solid arrow).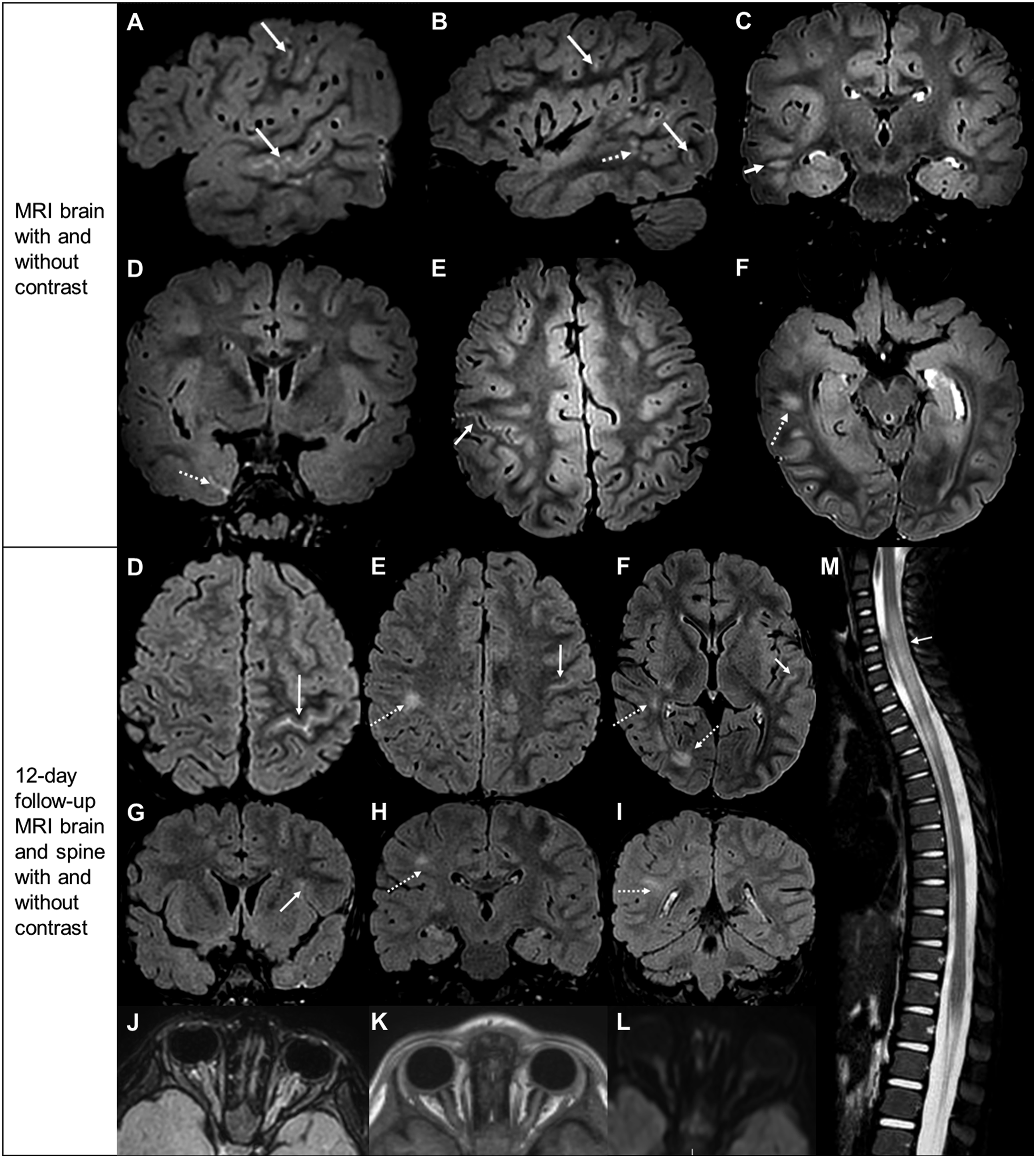
Unfortunately, he developed more frequent breakthrough seizures, at least once daily by the third week post-discharge. He became more irritable and developed worsening imbalance. He was readmitted to the hospital for further workup (Table 1). Fundus examination revealed left optic nerve edema. Brain MRI revealed new T2 hyperintensities in the optic nerves and right temporal and parietal subcortices and areas of leptomeningeal enhancement in the left central sulcus and temporal lobe (Figure 2G-L). Spine MRI showed a longitudinally extensive T2 hyperintensity of the central spinal cord from C4-T3 (Figure 2M), consistent with myelitis.
LP showed a normal opening pressure. CSF analyses was remarkable for elevated WBCs (51 cells/cm2) with neutrophilic pleocytosis and normal protein and glucose (Table 1). Continuous 2-day EEG revealed left centrotemporal slowing with left mesial frontal focal sharp waves. He was diagnosed with ADEM and received IV methylprednisolone and IVIg, with resolution of behavioral, vision, and gait disturbances after 5 days. Serum autoimmune panel was high-positive for anti-MOG antibody (1:1000 titer). He was discharged on oral corticosteroid taper with monthly IVIg.
Two weeks later, he developed loss of appetite and sleepiness and dragged his right leg while walking. High-dose corticosteroids were reinitiated, but he continued to have breakthrough seizures monthly, despite increasing oxcarbazepine to 540 mg (29 mg/kg) daily. MRI 3 months later showed resolution of signal abnormalities throughout the brain and spine. Despite this, his symptoms continued. Repeat serum anti-MOG antibody showed 1:100 titer. He received seven cycles of plasmapheresis with symptom resolution and was discharged with rituximab infusions. On 6-month follow-up, he remained seizure-free with the same dose of oxcarbazepine, and his gait was normal.
Discussion
The clinical manifestations of MOGAD vary in part by location of nervous system involvement and have included vision impairment, cranial nerve deficits, focal motor or sensory deficits, bladder or bowel dysfunction, and/or impaired coordination.3,4 Although our cases had distinct initial presentations, their diagnostic pathways were related, with hospital revisitations due to worsening symptoms despite initial measures, repeat neuroimaging revealing inflammatory signs, and subsequent broad serum and CSF workup with the detection of MOG-IgG. Similar brain MRI findings in both patients included ON, cortical/subcortical white matter lesions, and leptomeningeal contrast enhancement. Both patients had CSF pleocytosis with neutrophilia. The patients differed in treatment outcomes, as the second case with high anti-MOG antibody titer relapsed after plasmapheresis and required switching to rituximab for better control.
There have been few reports of MOGAD presenting with isolated ICH.5,6 In our first case, the identification of papilledema could have been due to ICH, bilateral ON, or both. If not fully worked up, particularly with CSF analysis or neuroimaging, this clinical presentation could be mistaken for idiopathic intracranial hypertension.7,8 When MOG antibody testing was unavailable, a 2015 cohort study found that one third of children with inflammatory demyelinating disease had elevated CSF opening pressures, thought to be due to increased antibodies and inflammatory cytokines disrupting the blood brain barrier and causing dysregulation of CSF flow dynamics. 9 We suspect that ICH with MOGAD may follow a similar pathophysiologic mechanism.
Our second patient presented with FLAIR-hyperintense lesions in anti-MOG associated encephalitis with seizures (FLAMES).10,11 Acute symptomatic seizures were found to occur in ∼20% of patients with MOGAD, supporting seizures as a presentation of MOGAD’s overlapping encephalitis-demyelination complex. 12 There was a reported case of relapse of seizures from FLAMES following corticosteroid taper, as in our case, suggesting that alternative immunosuppression, such as IVIg, rituximab, or tocilizumab, may be warranted. 11 Although our patient’s follow-up brain MRI showed diminished T2 hyperintensities, he remained symptomatic. In another case series, isolated new onset seizure with MOGAD was found to have a latency of up to 48 months before demyelination may be observed on MRI. 13 We suspect that nervous system involvement from MOGAD may not always be fully visualized on MRI.
The WBC differential within the CSF of both patients is also important to analyze. Both had elevated CSF WBCs, with abnormally elevated neutrophils. Patient 1 also had a normal proportion of lymphocytes and abnormal presence of eosinophils, whereas Patient 2 had neutrophilic predominance. In MOGAD, the presence of eosinophils and increase in neutrophils, in the absence of infections, are indicators of complement activation and indirectly an antibody-mediated disease. 14 In a study assessing CSF findings of 100 MOG-positive adults, neutrophils were present in 50% and more commonly associated with more acute attacks. 15
Our cases demonstrate the clinical variability and syndromic overlap in presentations of MOGAD, highlighting challenges associated with their initial diagnoses and value of early serial neuroimaging, specifically MRI of the brain, orbits, and spine with and without contrast. We also show differences in their management with immunotherapy based on their varied prognoses. As more cases of MOGAD unravel, we anticipate a continued growth of the clinical spectrum of reported cases demonstrating the versatility of MOGAD, the clinical chameleon.
Footnotes
Authors’ Note
The authors have obtained written informed consent from the father of the patient in Case 1 and mother of the patient in Case 2. These are available for verification upon request.
Acknowledgements
We would like to thank Dr. Morgan Harris (University of Iowa, Department of Pediatrics) for her assistance in initial literature review for the first case.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.