
Abstract
Acute hepatic porphyria (HP) often presents with recurrent neurovisceral symptoms in young adults, mimicking more common neurological conditions such as Guillain-Barré Syndrome (GBS) and posing significant diagnostic challenges. We report a case of a 25-year-old male who presented with progressive weakness of all 4 limbs over 4 days, culminating in respiratory paralysis requiring mechanical ventilation. Neurological examination revealed acute flaccid paralysis with areflexia, and nerve conduction studies showed acute motor axonal neuropathy, initially supporting a diagnosis of GBS. However, the patient’s young post-pubertal age, onset in the upper limb with proximal weakness, pure motor axonal neuropathy, and presence of hyponatremia due to the syndrome of inappropriate antidiuresis raised suspicion of acute HP. Screening with qualitative urine porphobilinogen testing, followed by quantitative confirmation, diagnosed acute HP. The patient was treated with intravenous dextrose in the absence of hemin, resulting in gradual clinical improvement. This case underscores the importance of distinguishing acute HP from GBS and the need for early recognition and screening to initiate life-saving therapy.
Keywords
Introduction
Porphyria remains a frequent source of diagnostic confusion due to its heterogeneous presentations. 1 Acute hepatic porphyria (HP) primarily affects young, post-pubertal adults. It typically presents as recurrent episodes of neurovisceral symptoms, which closely mimic more common neurological conditions such as Guillain-Barré syndrome (GBS).1-3 This clinical overlap leads to significant diagnostic challenges, necessitating a high index of suspicion. We present a case of acute HP initially misdiagnosed as GBS, underscoring the importance of prompt recognition, appropriate diagnostic testing, and immediate treatment.
Case Presentation
A 25-year-old male presented to the emergency medicine unit with progressive weakness of all 4 limbs over 4 days. The motor weakness had an acute onset, initially affecting the right upper limb and then rapidly progressing to the right lower, left lower, and finally the left upper limb within 2-3 days. The weakness was proximal at onset, causing difficulty in lifting his arm and getting up from a sitting position, before progressing distally, leading to grip weakness and slippage of footwear. One day before presentation, he developed difficulty breathing and speaking. His condition progressively deteriorated, and he was unable to speak in complete sentences, gasping for breath between words.
He did not experience difficulty swallowing, ptosis, visual disturbances, or diurnal symptom variation. There was no history of sensory disturbances, bowel or bladder dysfunction, back pain, band-like sensations, seizures, or altered sensorium. Additionally, there was no preceding history of fever, coryza, sore throat, rash, abdominal pain, diarrhea, vaccination, trauma, or falls. He had no history of similar past episodes, nor a family history of paralysis or unexplained neuropsychiatric illness.
On examination, he was conscious and oriented, with a pulse of 64 per min, blood pressure of 130/80 mmHg, and a respiratory rate of 28 per min, with rapid, shallow breathing. Neurological evaluation revealed decreased muscle tone in all 4 limbs, motor power graded 1/5 bilaterally, and areflexia with flexor plantar responses. Cranial nerve and sensory examinations were normal. Systemic examination was unremarkable.
Laboratory findings demonstrated hyponatremia (serum sodium 126 mmol/L), consistent with syndrome of inappropriate antidiuresis (SIAD), as evidenced by reduced serum osmolarity (274 mOsm/L), increased urine osmolarity (561 mOsm/L), elevated urinary sodium excretion (21 mmol/L), and normal adrenal and thyroid function tests. Liver and renal function tests were normal. Nerve conduction studies revealed acute motor axonal neuropathy. Lumbar puncture showed acellular cerebrospinal fluid with mildly elevated protein levels (70 mg/dL) and normal glucose (83 mg/dL). Contrast-enhanced magnetic resonance imaging of the brain and spine was unremarkable.
The patient required mechanical ventilation due to respiratory paralysis. Intravenous immunoglobulin (IVIg) was initiated for suspected GBS. However, screening for acute HP was performed using the Watson–Schwartz test, a qualitative spot urine test for porphobilinogen (PBG), which returned positive. Treatment was initiated with carbohydrate loading with intravenous dextrose (300 g/day), as hemin therapy was not available. Subsequent quantitative PBG testing confirmed elevated levels (3.3 μmol/mmol creatinine, normal <2.6), confirming a diagnosis of acute HP. SIAD was managed with fluid restriction. The patient gradually improved and was successfully weaned off ventilator support. He was educated on porphyria, including its known triggers, drugs to avoid, and the risk of recurrent attacks. At the 6-week follow-up, motor power improved to 4-/5.
Discussion
Porphyrias are metabolic disorders caused by deficiencies or increased activity of specific enzymes in the heme biosynthetic pathway. When these enzyme defects are physiologically significant, they lead to the overproduction of pathway precursors, which accumulate, enter the circulation, and are excreted in urine or bile. Porphyrias are broadly categorized into hepatic and erythropoietic types based on the site of metabolite accumulation. Clinically, they are further categorized into neurovisceral or cutaneous forms. 1
Among the 5 types of HPs, four—acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and δ-aminolevulinic acid (ALA) dehydratase deficiency porphyria (ADP)—present with acute neurovisceral symptoms. In contrast, porphyria cutanea tarda, the most common porphyria overall, manifests with blistering skin lesions. 1 Among the acute HPs, AIP, HCP, and VP are inherited as autosomal-dominant disorders, whereas ADP, a rare form, follows an autosomal-recessive inheritance pattern. AIP, HCP, and VP are characterized by elevated urine PBG levels, whereas ADP is distinguished by elevated urine ALA but normal urine PBG.1,2 Differentiating AIP from HCP and VP requires plasma and fecal porphyrin analysis as well as genetic testing for hydroxymethylbilane (HMB) synthase mutation; however, these tests are not routinely available and were not performed in this patient. Given that AIP is the most common acute HP, it remains the most likely diagnosis in this case. Importantly, the treatment of acute neurovisceral episodes in HCP and VP is similar to that of AIP.2-5
Differentiating Features Between GBS and Acute Hepatic Porphyria in Acute Flaccid Paralysis.

Abbreviations: AIDP- Acute inflammatory demyelinating polyneuropathy; AMAN- Acute motor axonal neuropathy, AMSAN- Acute motor-sensory axonal neuropathy, GBS- Guillain-Barré syndrome, HCP- Hereditary coproporphyria, SIAD- Syndrome of inappropriate diuresis, VP- Variegate porphyria.
Although acute HP is often triggered by drugs, fasting, or hormonal fluctuations, sporadic cases without an identifiable precipitant are well recognized, as illustrated by this patient. A lack of response to standard GBS treatments, such as IVIg, should also prompt consideration of alternative diagnoses, including acute HP. Given the overlapping clinical features between GBS and porphyric neuropathy, early screening with urine PBG testing is crucial in atypical cases (Table 1).7,10 In resource-limited settings, qualitative testing of fresh early morning spot urine samples over 3 consecutive days provides a practical bedside screening, with the Watson-Schwartz test yielding results within hours (Figure 1).
10
If any of the 3 samples test positive, early treatment should be initiated, particularly in severe cases. For confirmation, quantitative testing of the same urine sample is performed; however, turnaround time is often prolonged (a few days to a week), depending on laboratory availability. A positive result confirms acute HP, guiding further treatment, while a negative result warrants a reassessment of clinical suspicion and consideration of alternative diagnoses.
10
Suggested algorithm for diagnosing acute HP in acute flaccid paralysis. Abbreviations: GBS- Guillain-Barré syndrome, HP- Hepatic porphyria, PBG- Porphobilinogen.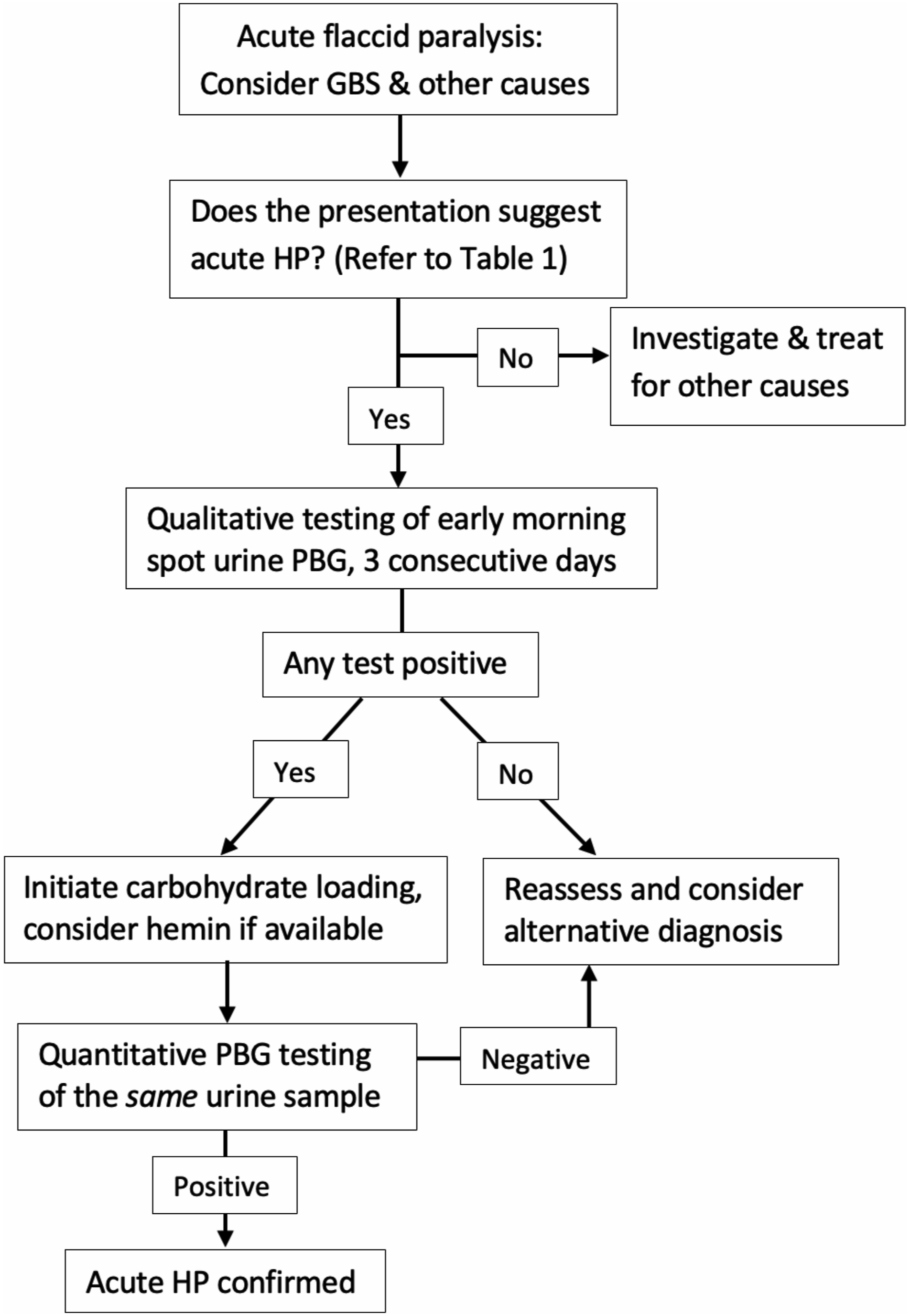
Intravenous hemin is the first-line therapy for neurovisceral attacks of acute HP; however, its high cost and limited availability can pose significant challenges.1-5 In such cases, carbohydrate loading with intravenous dextrose (at least 300 g daily) is an effective alternative treatment.1,7,10
Conclusion
This case underscores the diagnostic challenge of acute HP masquerading as GBS. Early recognition is critical, as delays in diagnosis and treatment can lead to severe, potentially life-threatening complications. Clinicians should maintain a high index of suspicion for acute HP in young patients presenting with acute flaccid paralysis, especially when features such as pure motor axonal neuropathy, hyponatremia (SIAD), early autonomic dysfunction, or poor response to GBS-specific therapies are present. Spot urine PBG testing is a simple, rapid, and effective screening tool in resource-limited settings. In the absence of hemin therapy, prompt carbohydrate loading should be initiated to mitigate disease progression and improve outcomes.
Footnotes
Author Contributions
Kumar Porakapalli Yuvasai: patient management, collected patient data, drafted the manuscript. Sanveer Singh, Pratiksha Jayant Pai, Sathvik Reddy Erla: patient management, collected patient data. Ashok Kumar Pannu: patient management, collected patient data, drafted and revised the manuscript. The corresponding author is responsible for ensuring that the descriptions are accurate and agreed upon by all authors
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.