
Abstract
Keywords
Introduction
New-onset refractory status epilepticus (NORSE) is a rare and challenging clinical syndrome characterized by the sudden onset of refractory status epilepticus (RSE) in individuals without a prior history of seizures or identifiable acute structural, toxic, or metabolic causes. 1 The diagnosis of NORSE is typically considered after 48-72 hours of persistent seizures, allowing time for a comprehensive evaluation to rule out other causes. The incidence and prevalence of NORSE are difficult to ascertain accurately, likely due to its underdiagnosis and the absence of a standardized definition until recently. 2 However, it is estimated that approximately 0.3-2.4 per 100,000 individuals with seizures, or 10-20% of RSE cases, could be classified as NORSE. 3 The most common causes of NORSE are autoimmune and infectious encephalitis, with viral infections being the most frequent infectious trigger. However, in approximately 50% of cases, no specific etiology is identified, and the condition is classified as cryptogenic NORSE. 4 Cryptogenic NORSE presents a greater challenge, as the absence of a clear autoimmune or infectious cause limits the use of targeted therapies that could modulate disease activity. It is hypothesized that an immune-mediated inflammatory response underlies most cases of NORSE, which is why immunotherapy is often attempted, even in cryptogenic cases, with varying success. Recent consensus recommendation for acute treatment typically involves first-line agents like corticosteroids or intravenous immunoglobulins (IVIG), within 72 hours of onset of SE, although therapeutic plasmas exchange (TPE) was mentioned but not included in consensus, and second-line therapies such as tocilizumab, rituximab, cyclophosphamide, and anakinra. 2
For patients where no treatment proves effective, prolonged burst suppression may be necessary, often leading to an uncertain prognosis and consideration of care withdrawal due to the lack of alternative treatments. In such challenging cases, off-label and experimental therapies might be considered, especially when conventional options have been exhausted. This article presents a case of cryptogenic NORSE that was resistant to multiple antiseizure medications, IV anesthetics, and immunosuppression. The patient’s condition eventually improved with the use of IV ganaxolone and electroconvulsive therapy (ECT), leading to seizure cessation, recovery of consciousness, and a favorable outcome after 8 months of hospitalization.
Case Description
A 23-year-old woman with history of vitiligo presented 1 week after a recent febrile illness with new onset of generalized tonic-clonic seizures which did not respond to pre-hospital administration of benzodiazepines (Figure 1). She then received loading doses of intravenous (IV) levetiracetam and fosphenytoin, achieving temporary seizure cessation. Convulsive status epilepticus recurred requiring invasive mechanical ventilation and administration of IV anesthetics (midazolam, propofol then ketamine) to control ongoing seizures. During the attempt to wean the patient off propofol and midazolam infusions, she experienced recurrent seizures. To facilitate the weaning process, additional antiseizure medications, including lacosamide, clobazam, valproic acid, and perampanel, were administered. Despite these measures, the patient continued to experience super-refractory status epilepticus (SRSE), prompting the initiation of a pentobarbital infusion. This achieved adequate burst suppression, allowing for the successful discontinuation of propofol and midazolam by day 7. However, the patient continued to require ketamine in conjunction with pentobarbital to maintain seizure control. (A) Patient’s EEG on Admission. Continuous EEG on Admission Showing Bilateral Independent Rhythmic Spikes and Sharp Waves With Frequency 3-4 Hz. Similar Pattern Emerged Upon Attempts to Wean Sedation Throughout the Hospital Course. (B) MRI of the Brain. Axial FLAIR MRI Showing Slight Enlargement and Hyperintensity of the Right Hippocampus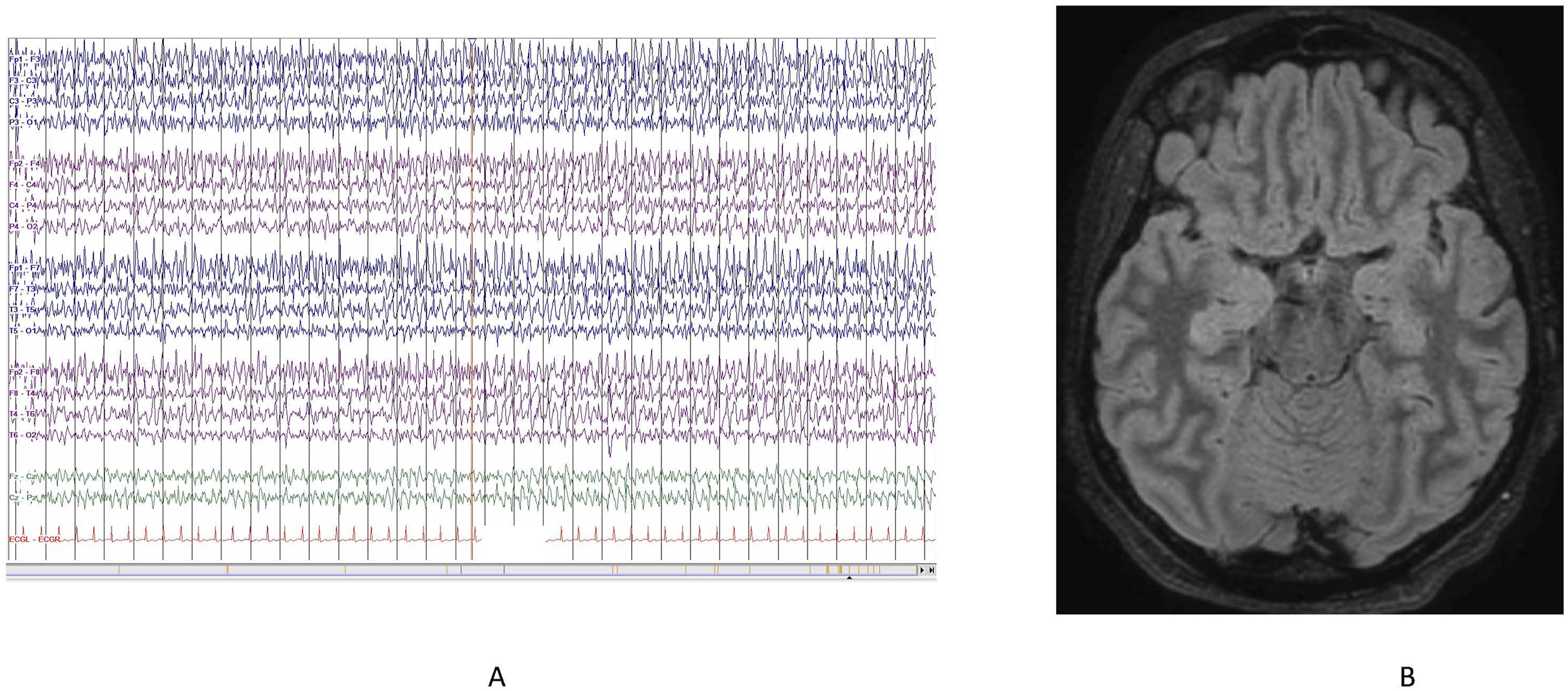
The patient underwent an extensive diagnostic workup. Cerebrospinal fluid (CSF) analysis revealed negative results for infectious meningitis/encephalitis PCR panel, autoimmune encephalitis, paraneoplastic panel, cytokine panel, viral studies (including HSV, EBV, Enterovirus, CMV, and West Nile), Lyme disease, VDRL, NMDA receptor antibody, cytology, and bacterial and fungal cultures. Serum studies were notable for positive lupus anticoagulant in two tests conducted 3 months apart. However, other markers for autoimmune conditions, including ANA/dsDNA, lupus, topoisomerase antibody, centromere antibody, RNA polymerase III antibody, GAD antibody, smooth muscle antibody, SLA autoimmune antibody, mitochondrial antibody, ceruloplasmin, LKM-1 microsomal IgG, C3/4, ENA, cardiolipin, and beta-2 glycoprotein IgG/M, were negative. Urine organic panel reveals only slight elevation of 5-oxo-proline, 4OH-phenylacetic acid and glyceric acid. Brain MRI with and without contrast showed an increased FLAIR signal and slight enlargement of the right hippocampus (Figure 1B).
Given concerns about an underlying inflammatory process, immunomodulatory therapies were promptly initiated. IV methylprednisolone was started on day 4 and continued for 5 days, followed by intravenous immunoglobulin (IVIG) on day 5 at a total dose of 2 g/kg. Ketogenic diet was introduced on day 14; however, persistent ketosis was not achieved. Concurrently, the patient received anakinra, an interleukin-1 receptor antagonist, at 100 mg daily for two weeks without success. Subsequently, tocilizumab, an interleukin-6 receptor antibody, was administered in four doses (4 mg/kg for the first two doses, 8 mg/kg for the third, and 12 mg/kg for the fourth), but no improvement was observed. IV rituximab was then administered, also without immediate benefit. During this period, the patient also underwent 5 sessions of plasmapheresis. Unfortunately, none of these treatments enabled the weaning of the barbiturate infusion. Given persistent coma and prolonged hospitalization, she underwent tracheostomy and percutaneous endoscopic gastrostomy. Timeline of Antiepileptic Therapy Administration During Hospital Stay in a Patient Cryptogenic NORSE Over the Span of 244 days. PLEX: Plasma Exchange. ECT: Electroconvulsive Therapy. K diet: ketogenic diet. MTPRED: Methyl Prednisone. VPA: Valproic Acid. PB: Phenobarbital. PTB gtt: Pentobarbital Infusion. KET gtt: ketamine Infusion. VER gtt: Midazolam Infusion. PRO gtt: propofol Infusion. FOS: Fosphenytoin. GNX: Ganaxolone. PGB: Pregabalin. OXC: Oxcarbazepine. CZP: Carbamazepine. TPM: Topiramate. PER: Perampanel. CLB: Clobazam. LCM: Lacosamide. LEV: Levetiracetam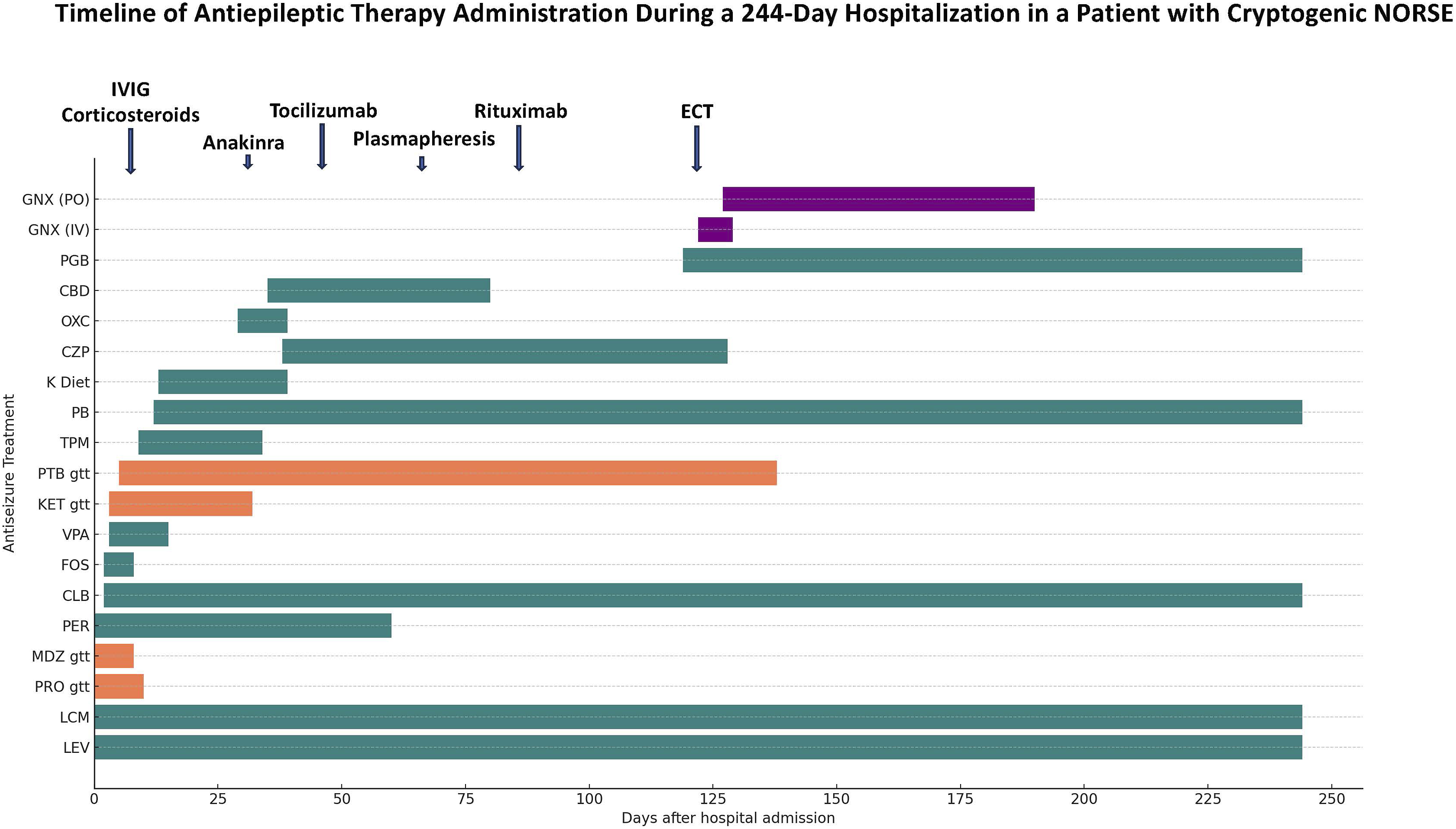
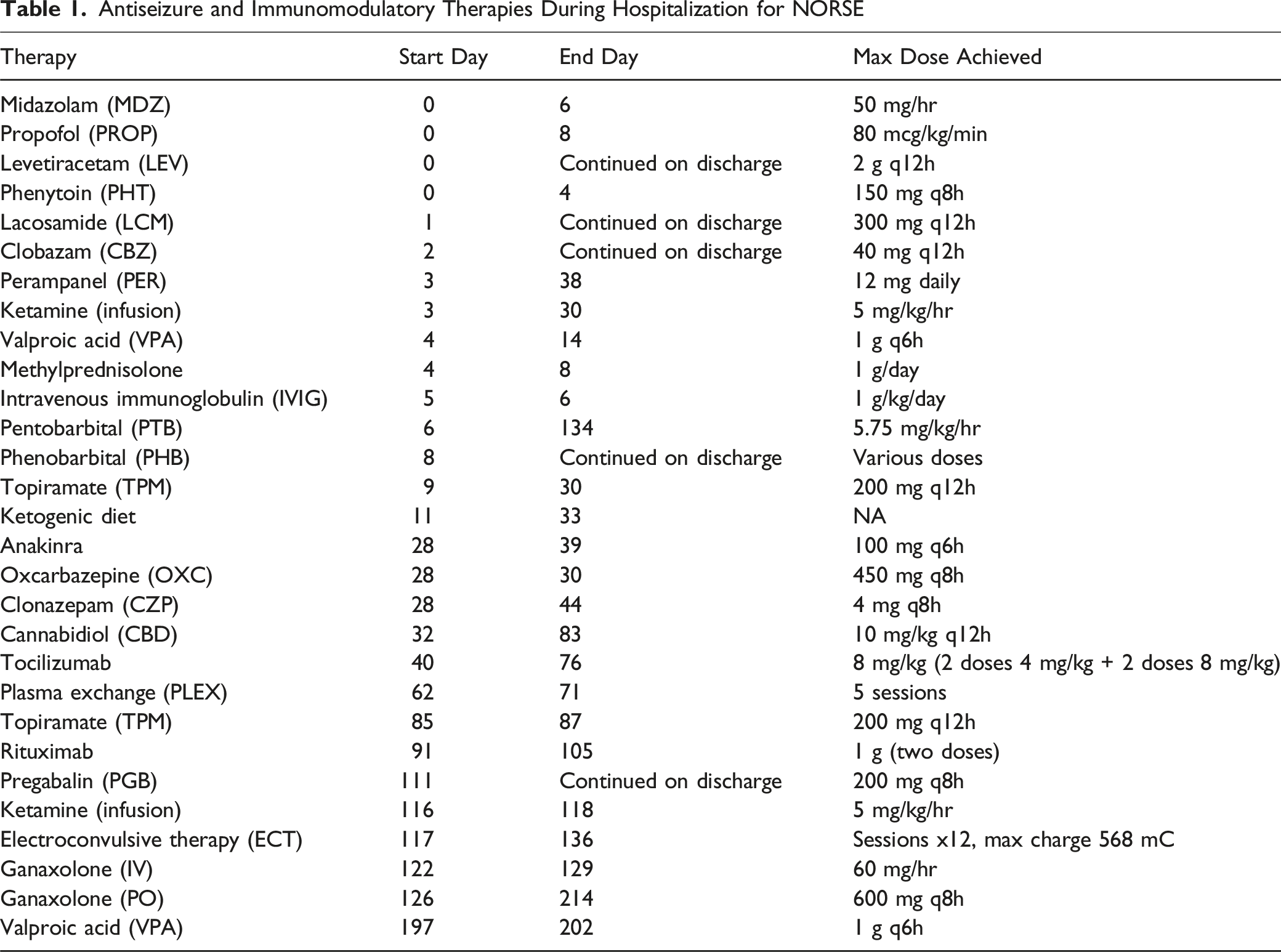
Given the protracted course of recurrent SRSE and the inability to wean the patient from pentobarbital, we submitted a request for an emergency single patient access IND (eIND) from the FDA for ganaxolone, a GABAA receptor modulator developed by Marinus Pharmaceuticals, for use in SRSE. While awaiting eIND approval and the delivery of the medication, ECT was initiated. ECT began on day 117 of the hospital stay, targeting the bilateral temporal lobes with targeting response of any seizure. All IV anesthetics and PTB were held prior to the ECT procedure to allow for development of seizures induced by ECT. (see the clinical and electrographic response to each ECT session in Supplemental Table.) IV ganaxolone was started 6 days after ECT initiated, administered over a total of 7 days. The dosing regimen included a 20 mg bolus, followed by an infusion at 60 mg/hr for 60 minutes, and then 42 mg/hr for 5 days, with tapering completed by day 7. Following IV treatment, ganaxolone was transitioned to enteral administration at a dose of 600 mg every 8 hours. During this time, the patient underwent a total of 12 ECT sessions over the course of 17 days. Nine days after the initiation of IV ganaxolone, the pentobarbital infusion was successfully weaned off (Table 1 and Figure 2).
During and after the initiation of ganaxolone, continuous EEG monitoring during showed improvement, manifesting as increased inter-burst intervals and attenuation in the amplitude and frequency of epileptiform discharges, allowing to wean pentobarbital to completely off, followed by gradual re-emergence of organized posterior dominant rhythm. Following pentobarbital discontinuation, she demonstrated gradual recovery of consciousness, with consistent response to simple commands within 1-2 weeks; the tracheostomy was subsequently decannulated. However, she continued experienced occasional brief electroclinical seizures lasting 10-30 seconds, these were managed by adjusting the phenobarbital dosage. The dose was carefully titrated to balance seizure control while preserving her mental status. Enteral ganaxolone was gradually weaned off according to the company protocol, with the tapering process completed by 90 days. For the remainder of her hospital stay, the patient was completely weaned off enteral ganaxolone and demonstrated significant improvement in her mental status, with only occasional clinical seizures. By the time of discharge, she was awake, oriented, and able to follow complex commands and engage in normal conversation. However, she continued to experience memory deficits and anxiety and had developed critical illness neuropathy and myopathy, leading to severe lower extremity weakness, necessitating the use of a wheelchair for mobility. She was eventually discharged to a rehabilitation facility on day 244 after the onset of her seizures. At the time of discharge, the patient’s antiseizure medications included clobazam 20 mg q12h, lacosamide 300 mg q12h, levetiracetam 2g q12h, pregabalin 200 mg q8h and phenobarbital 113.4 mg q6h.
At the 6-month follow-up, the patient was awake, alert, and able to engage in normal conversation. She can move her upper extremities against gravity and is able to stand and walk with assistance. Her Glasgow Outcome Scale–Extended (GOSE) score was 7, corresponding to a Lower Good Recovery — able to resume normal activities with some injury-related problems, primarily limited by severe weakness from the prolonged hospital stay. While the patient can live independently and return to some daily activities, she continues to experience significant limitations that affect her ability to fully resume her pre-illness lifestyle, including challenges in work or social functioning. Her Barthel Index score was 70, reflecting a moderate degree of independence in activities of daily living, though still requiring some assistance.
Discussion
We report the case of a young woman with no prior medical history who was admitted for new onset SRSE following an upper respiratory viral infection, likely representing febrile infection-related epilepsy syndrome (FIRES). Despite a comprehensive workup, the etiology remained unclear, leading to a diagnosis of cryptogenic NORSE. NORSE is a challenging clinical entity with an in-hospital mortality rate as high as 40-54% and poor response to conventional antiseizure medications 5 ; approximately one-third of patients requires multiple intravenous anesthetics for prolonged periods to achieve complete seizure cessation and/or burst suppression. International consensus guidelines recommend early immunotherapy and ketogenic diet for treatment of cryptogenic NORSE, followed by second-line immunomodulators in cases refractory to initial therapy. 6
In our patient, first-line immunotherapies (IVIG, plasmapheresis, corticosteroids) and second-line agents (anakinra, tocilizumab, rituximab) failed to produce clinical improvement, even after sufficient time was allowed for these treatments to take effect. In such cases, the next steps in management are uncertain, and the prolonged hospital course with ongoing status epilepticus often necessitates difficult discussions about goals of care and the potential withdrawal of life support in a young, previously healthy patient. However, our case demonstrates that even in severe cases of cryptogenic NORSE, recovery of consciousness and a reasonably favorable outcome could still occur. The concurrent use of IV ganaxolone and ECT likely played a crucial role in achieving seizure cessation, though further evidence is required to validate this approach.
Given the limited evidence supporting the use of second-line immunomodulators, it is common practice to trial multiple agents, as was done in our case. Anakinra is a recombinant IL-1 receptor antagonist. In a review of 25 pediatric patients with Febrile Infection-Related Epilepsy Syndrome (FIRES) treated with anakinra, 11 patients experienced a >50% reduction in seizures within the first week of treatment. 7 Tocilizumab, a humanized monoclonal antibody targeting the IL-6 receptor, has shown potential in treating cryptogenic NORSE. Donnelly et al reported a case involving a young female patient with cryptogenic NORSE who did not respond to conventional immunosuppressive and immunomodulating therapies; however, her condition improved following two doses of 300 mg tocilizumab infusion. 8 Rituximab is a monoclonal antibody that specifically targets the CD20 antigen on B cells. Case reports of patients with refractory status epilepticus (RSE) associated with serum antibodies such as NMDAR, LGI1, or VGCC suggest that treatment with rituximab can result in sustained seizure control and significant clinical improvement. 9 However, our patient did not respond to rituximab either.
ECT, historically used in epilepsy since the 1930s but now rarely employed, was initiated as salvage therapy. It involves delivering electrical stimulation to the brain under mild anesthesia, inducing a brief seizure. Its main side effect is reversible memory impairment; otherwise, it is considered effective and safe for refractory mood disorders and schizophrenia. The first reported use of ECT for status epilepticus was by Viparelli et al in 1992, describing a 19-year-old woman with refractory partial seizures unresponsive to intravenous diazepam and enteral phenytoin. She received two ECT sessions 48 hours apart; the first reduced seizure frequency, and the second achieved complete cessation. 10 In a scoping review by Dowd et al (16 studies; 40 SRSE patients), ECT terminated seizures in ∼80% of adults and 100% of children before discharge. 11 In our case, the patient received her first ECT treatment 18 weeks after the onset of her seizure, with a charge of 568 mC. Over 17 days, she underwent a total of 12 sessions. Upon the patient’s recovery, she experienced memory issues; however, it remains unclear whether these cognitive deficits were a consequence of the ECT or prolonged status epilepticus.
Allopregnanolone is a potent, endogenously produced neuroactive steroid that modulates multiple GABAA receptor subtypes and plays a critical role in regulating cortical excitability. It has demonstrated significant antiseizure effects in various animal models. In a recent small open-label cohort study investigating brexanolone, an analog of allopregnanolone, for the treatment of SRSE, its use was associated with a high rate of successful weaning from third-line anesthetic agents. 12 Ganaxolone, a 3-beta-methylated synthetic analogue of allopregnanolone, is a novel positive allosteric modulator of GABAA receptors. It modulates synaptic receptors (comprising alpha and gamma subunits) and, unlike benzodiazepines, also modulates extrasynpatic GABAA receptors (comprising alpha and delta subunits), leads to persistent conductance and dampening of network excitability. In its enteral form, it was approved by FDA for treatment of seizures associated with cyclin-dependent kinase-like 5 (CDKL5) deficiency disorder (CDD) in patients 2 years of age and older. 13 In an open-label, phase 2 trial using the IV formulation involving 17 patients with refractory seizures—who had failed to respond to at least 1 second-line IV seizure medication—none required escalation to third-line IV anesthetics within the 24 hours following the initiation of ganaxolone. 14 In a recent meta-analysis, enteral ganaxolone was shown to reduce seizure frequency by ≥ 50% and had an acceptable tolerability and safety profiles. 15 IV ganaxolone is not currently commercially available but has been recently studied in the RAISE trial (RAndomized Therapy In Status Epilepticus) for RSE. 16 RAISE was a phase 3, double-blind, randomized, placebo-controlled trial that evaluated the safety and efficacy of IV ganaxolone for treating refractory status epilepticus in patients who had failed at least two prior antiseizure medications. Patients were randomized to receive either IV ganaxolone or placebo, in addition to standard of care treatment. The intent-to-treat population consisted of 96 patients, with 49 in the ganaxolone arm and 47 in the placebo arm. The trial successfully met its first co-primary endpoint, demonstrating a statistically significant proportion of patients achieving cessation of status epilepticus within 30 minutes of initiating IV ganaxolone compared to placebo (80% vs 13%, P < 0.0001). However, the trial did not meet the second co-primary endpoint; there was no statistically significant difference in the proportion of patients who did not progress to IV anesthesia within 36 hours post-treatment (63% vs 51%, P = 0.162), Adverse events were comparable between the treatment and placebo groups, though hypotension was more frequently observed in the ganaxolone arm. Interestingly, preliminary EEG analysis suggested that patients receiving ganaxolone had a durable reduction in seizure burden through 36 hours, with an 88% median reduction in seizure activity compared to 38% in the placebo group, indicating that progression to IV anesthesia may have been influenced by factors other than seizure control alone. 16 Since the co-primary endpoint of avoiding progression to IV anesthesia within 36 hours was not achieved, efficacy remains unproven. Thus, IV ganaxolone remains investigational and warrants further study.
Antiseizure and Immunomodulatory Therapies During Hospitalization for NORSE
Conclusions
NORSE remains a challenging condition with no proven effective treatments, particularly given its high in-hospital mortality rate. In this case, despite multiple prolonged trials of antiseizure medications, the patient’s SRSE ultimately resolved following the combined initiation of IV ganaxolone and ECT. While clinical improvement was temporally associated with ganaxolone administration, the close timing of ECT makes it difficult to definitively attribute the response to either intervention alone. It remains possible that ECT played a significant role, or that the therapeutic effect resulted from the combination of both treatments. This case highlights the need for further investigation into the individual and combined roles of IV ganaxolone and ECT in managing refractory status epilepticus.
Supplemental Material
Supplemental Material - Successful Treatment of Cryptogenic NORSE Resistant to Immunosuppression With Intravenous Ganaxolone and Electroconvulsive Therapy
Supplemental Material for Successful Treatment of Cryptogenic NORSE Resistant to Immunosuppression With Intravenous Ganaxolone and Electroconvulsive Therapy by Zheng D. Lan, Rishi Malhotra, Ali Naqvi
Footnotes
Acknowledgement
We would like to acknowledge Dr Zheng Lan, our colleague and neurocritical care fellow, who sadly passed away during the revision of this manuscript. Dr Lan was a dedicated and hard-working physician whose commitment to patient care and academic inquiry was deeply valued by all who worked with him. We are grateful for his contributions to this work and to the field at large. We extend our heartfelt condolences to his family, friends, and colleagues.
Ethical Considerations
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Consent to Participate
Informed consent was obtained from the patient’s guardian for the purpose of publication.
Author’s Contributions
I.M. and R.M. conceived the treatment plan. Z.L., IM collected the data. Z.L. and I.M. wrote the manuscript. R.M. and A.N. also reviewed and edited the manuscript. H.V. and M.B. provided legal and logistical support and reviewed the manuscript for potential conflicts of interest, as representatives of the company supplying the investigational drug.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.B. and H.V. are employees of Marinus Pharmaceuticals, Inc and hold shares in the company. The remaining authors have no conflicts of interest, received no compensation from Marinus Pharmaceuticals, and disclose no financial relationships with the company.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.