
Abstract
Leucine-rich glioma-inactivated-1 (LGI1) IgG-associated autoimmune encephalitis typically presents with faciobrachial dystonic seizures, hyponatremia, and temporal lobe MRI abnormalities. However, the clinical spectrum of LGI1 IgG-associated AE extends beyond these classical features, with reports of diverse seizure semiologies and atypical presentations challenging traditional diagnostic paradigms. This study reports a male in his late 60s presenting with recurrent episodes of right foot cramping and electric shock-like pain, progressing to generalized tonic-clonic seizures. Initial workup suggested chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), but LGI1 IgG antibodies were subsequently detected. Despite treatment with intravenous immunoglobulin (IVIG), the seizures persisted. This case highlights the heterogeneity in LGI1 encephalitis presentations. The patient’s proprioceptive-induced reflex seizures represent a rare manifestation of the disease. Treatment with rituximab, a B-cell depleting therapy, resulted in significant improvement, suggesting its potential effectiveness in LGI1 encephalitis with a resistant seizure course. The case also emphasizes the intricate interplay between the mesial temporal lobe, basal ganglia, and frontal regions in LGI1 encephalitis pathophysiology.
Introduction
Leucine-rich glioma-inactivated-1 (LGI1) IgG-associated autoimmune encephalitis is increasingly recognized as a cause for late onset seizure, typically affecting older adults and presents with faciobrachial dystonic seizures (FBDS), cognitive changes & behavioral changes and hyponatremia. 1 Early diagnosis and immunotherapy is critical as timely intervention can prevent irreversible hippocampal damage and drug resistant epilepsy. Although FBDS are most commonly reported seizure, multiple studies have shown that LGI 1 antibody encephalitis can present with broader semiologies including focal motor seizure, sensory seizure, generalized tonic clonic seizure, complicating early recognition of disease.2,3 We report an unusual case of LGI1 antibody associated autoimmune encephalitis in a man in his late 60s initially presented with proprioceptive induced reflex focal motor seizure that secondary generalized. This case emphasizes the importance of recognizing the diverse seizure semiology associated with LGI1 encephalitis and considering an autoimmune etiology in patients whose seizures remain refractory to standard antiseizure medications.
Case Presentation
A male in his late 60s with a history of prostate cancer and prediabetes was admitted to the emergency department in late 2019 with two consecutive episodes of generalized tonic clonic seizures (GTCS). Upon regaining consciousness, the patient reported a month-long series of episodic right foot cramping and electric shock-like pain. At times, this sensation would spread to his hip, rendering him unable to move the lower half of his body, yet his awareness remained intact. During several instances, the sensation would move further up, leading to full-body shaking, loss of consciousness, bowel incontinence, tongue biting, and postictal confusion. The patient had experienced multiple similar episodes for a month prior to the admission. During the physical examination, notable findings included a mild decrease of vibration sensation distally in both hands and the left lower extremity. However, the right lower extremity displayed a significant reduction in the same modality, with a complete loss of proprioception in the right great toe. Initial magnetic resonance imaging (MRI) of the brain did not show any abnormalities. Due to the patient’s presentation and the progression to GTCS, he was admitted to the epilepsy monitoring unit (EMU) for 3 days that did not capture any significant event. He was started on Levetiracetam 750 mg BID which proved ineffective. Later, he was switched to oxcarbazepine 600 mg BID but it was discontinued due to an allergic reaction. Subsequently, he was restarted on levetiracetam 1000 mg with addition of clobazam 20 mg BID.
Two months after his initial presentation, the patient sought evaluation from an outside neuromuscular specialist for comprehensive evaluation of right foot cramping and “electric shock” type of pain. The electrodiagnostic study performed at outside facility was interpreted as sensorimotor patchy demyelinating polyneuropathy suggestive of early chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) contributing to neuromyotonia. Intravenous immunoglobulin (IVIG) 2 g/kg divided over 5 days was initiated and continued every 2 weeks, in the context of presumed CIDP. Steroids were not pursued due to prediabetic status. However, by the third cycle of IVIG, the patient had experienced more than 8 episodes of GTCS and 80–90 episodes of right foot cramping, now more indicative of focal motor seizures. Subsequent workup revealed positive LGI1 IgG antibody in serum (cell based assay, Euroimmun Lubek), CASPAR2 was negative. Cerebrospinal fluid (CSF) studies were considered but not performed, due to patient denial.
Almost 8 months after onset of symptoms, the patient continued experiencing worsening seizures, alongside mood changes and memory deficits. Subsequent MRI scans revealed interval progression from normal to right hippocampal atrophy with mild T2/FLAIR hyperintensity without any contrast enhancement (Figure 1). A repeat electrodiagnostic testing at our facility did not show any evidence for neuromyotonia or demyelinating neuropathy. Given the lack of favorable response to IVIG, plasma exchange therapy (PLEX) was initiated. Despite five rounds of PLEX and continued compliance with anti-seizure medications (ASMs), he continued to have focal motor seizures and GTCS events. Right hippocampal atrophy with mild T2/FLAIR hyperintensity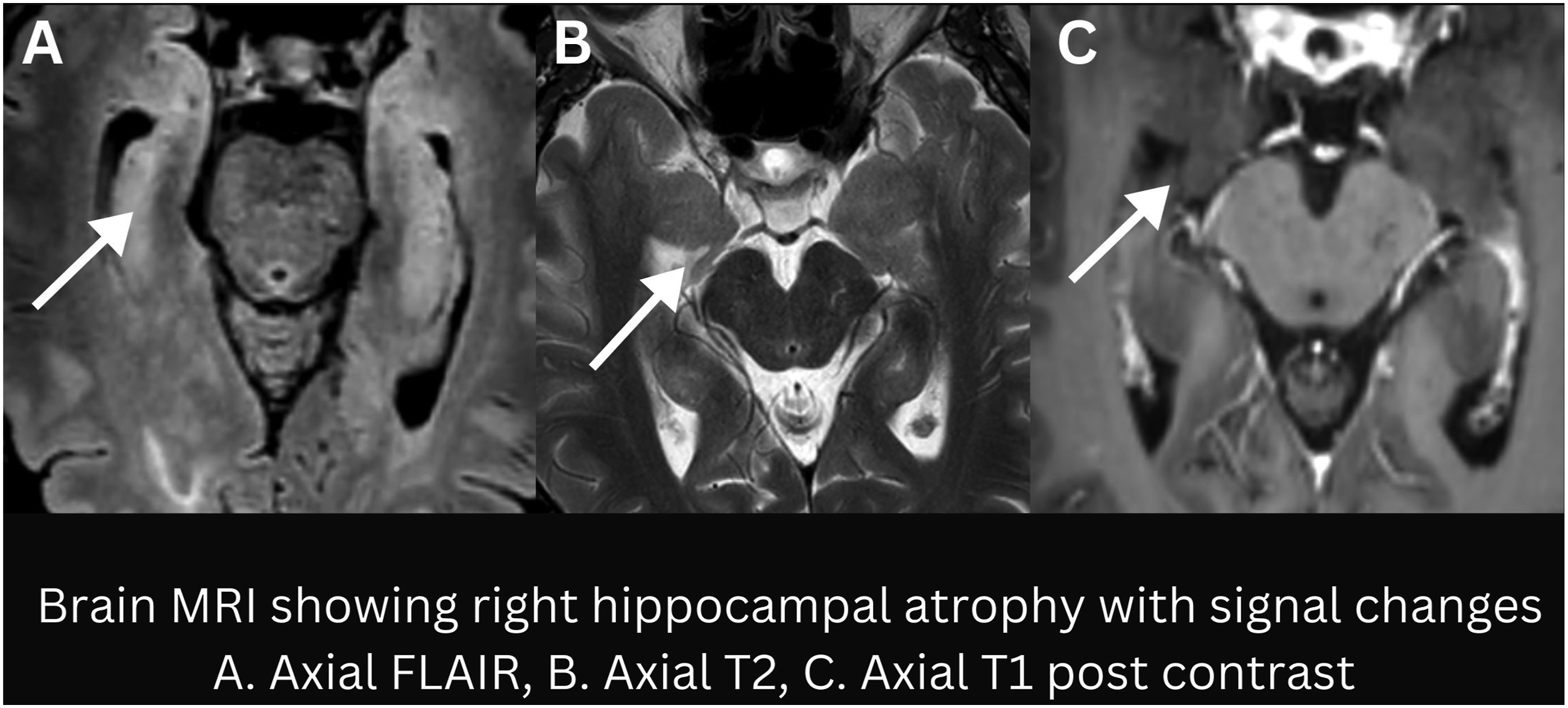
The patient was readmitted to EMU and his ASMs were stopped. The patient mentioned that most of his seizures start in the right foot, either they will start after spontaneous flexion or with voluntary flexion of the right ankle to the extent that he was afraid to get out of the bed. One seizure was captured on day 3 of admission, starting with a tingling/twitching sensation between the big toe and second toe of the right foot. This progressed to a spasm of the right leg seen as a flexion at the knee and hip, ultimately resulting in bilateral asymmetric tonic seizure followed by generalization of seizure. The patient was able to answer questions until secondary generalization. The second seizure on day 4 of admission, was similar in progression but started with voluntary flexion at the ankle in the right leg (Video 1 at 0.15 sec). Both seizures had unclear onset, with the second one showing paroxysmal alpha activity in the bifrontal region 9 sec after foot dorsiflexion suggesting focal clinical onset rapidly progressing to generalized tonic clonic seizure (Figure 2A and 2B). Paroxysmal alpha activity in the bifrontal region progressing to generalized tonic clonic seizure
The patient was initiated on rituximab 1 gm intravenous infusion followed by another 2 weeks later. The anti-seizure regimen was changed to 200 mg BID lacosamide. Following rituximab and lacosamide initiation, no further seizure could be triggered by leg movements, including active or passive flexion of the right foot. A year after his initial presentation, the patient was doing well on lacosamide and rituximab infusion every 6 months. Remarkably, a year later in January 2022, seizure freedom was maintained, furthermore cognitive and neuropsychiatric deficits resolved completely. In January 2023, the interval between rituximab infusions spaced out to 8 months as he maintained symptom free status and stable MRI.
Discussion
The typical clinical picture for LGI1 autoimmune encephalitis involves an older male (> 60 years) presenting with faciobrachial dystonic seizures (FBDS), hyponatremia, and increased T2/FLAIR medial temporal lobe signal on brain MRI. 1 While serum LGI1 antibodies detected by cell based assay are considered highly specific, the absence of CSF profile in this case, along with a clinical presentation of proprioceptive stimuli induced focal seizures with secondary generalization, and focal motor seizure mimicking a neuromuscular disorder (CIDP vs neuromyotonia) created diagnostic ambiguity and delay in recognizing autoimmune encephalitic process. . Notably, Teng et al.’s review found that 68% of patients with LGI1 antibodies exhibited seizures beyond the typical FBDS. 2 Similarly, Tillman et al.’s study reported that 22% of their LGI1 antibody-positive patients experienced bilateral tonic-clonic seizures, further demonstrating the potential for generalized manifestations. 3
To better understand the unique presentation in this case, it is essential to delve into reflex seizures, a category of epilepsy where seizures are triggered by specific stimuli. These seizures can be induced by various external or internal sensory modalities, such as visual, auditory, somatosensory, or cognitive stimuli. 4 Our patient’s condition appeared to be consistent with proprioceptive stimuli-induced reflex seizures (PIRS), elicited by active or passive movements of the body or limbs, with some spontaneous events, indicating reflex facilitation rather than pure epilepsy syndrome.4,5 Previous studies have indicated potential roles of the basal ganglia, frontal cortex, and supplementary motor area in PIRS. Lishman et al.’s case series proposed a role for the basal ganglia in the initial tonic spasm, with possible spread through cortico-basal ganglia circuits. 4 Falconer et al.’s report highlighted localized cortical dysfunction, with seizure cessation observed after resection of scar tissue in the left frontal area near the motor cortex. 5 Pierelli et al.’s SPECT findings supported the supplementary motor area’s involvement, revealing regional hypoperfusion in the anterior right hemisphere. 6 Giovanni et al. further implicated this area by describing a PIRS case associated with a dysembryoplastic neuroepithelial tumor in the right supplementary motor area. 7 However, our case presents a unique constellation of features that challenges traditional understandings of PIRS. The presence of right mesial temporal lobe atrophy, T2/FLAIR changes, positive LGI1 antibodies, and secondary generalization with impaired awareness suggests a more intricate interplay of factors.
LGI1 antibodies work by altering AMPA receptor clustering resulting in neuronal hyperexcitability, particularly affecting the limbic system. 8 We propose that the initial trigger is proprioceptive input from ankle and knee flexion, lowering seizure threshold in perirolandic cortex through already disrupted inhibitory circuits, mediated by LGI1 antibodies. This heightened excitability likely spreads through cortico-basal ganglia loops, resulting in the characteristic initial limb spasm. Later, the involvement of additional areas, indicated by the right mesial temporal lobe atrophy and observed bifrontal epileptic discharge, contributed in seizure generalization and impaired awareness, consistent with LGI1 associated network dysfunction. Previously, Navarro et al’s study also reported that in 37% of their LGI1 antibody positive patients, tonic dystonic seizure propagated into frontal lobe seizures. Furthermore, they observed hypermetabolism in the mesial temporal lobe and primary motor cortex, occurring in parallel to the striatum, highlighting their involvement in LGI1 encephalitis. 9 Similarly, the prolonged diagnostic delay also reflects evolving disease processes where early sensorimotor phenomena likely characterize cortical hyperexcitability and later stages consistent with limbic involvement including cognitive changes and hippocampal atrophy. This temporal progression raises the possibility that LGI1 autoimmune encephalitis may initially cause focal cortical involvement before evolving into limbic encephalitis, leading to delay in diagnosis.
Although a variety of immunotherapies have been reported to be associated with favorable outcomes, there are currently no treatment guidelines for optimal management of LGI1 IgG-associated autoimmune encephalitis. A randomized controlled trial involving 17 patients with LGI1 or contactin-associated protein-like 2 (CASPR2) IgG autoimmune encephalitis showed that IVIG is more effective than a placebo in reducing seizure frequency. 10 However, even in this clinical trial, only a minority of cases achieved seizure freedom. In our case, due to ongoing seizures, new cognitive deficits and refractoryness to first line treatment prompted for rituximab administration resulting in rapid clinical improvement, including seizure cessation and cognitive recovery. Rituximab, mycophenolate and cyclophosphamide are commonly utilized second-line therapies for autoimmune encephalitis treatment. Rituximab was chosen for this patient because B-cell depleting therapies have demonstrated efficacy in antibody-mediated autoimmune encephalitis, and in terms of toxicity profile, rituximab is relatively well tolerated. 11
Understanding the precise mechanisms by which LGI1 antibodies influence network dynamics and seizure propagation pathways, particularly the specific interaction with the mesial temporal lobe, basal ganglia and frontal regions, remains an important area for future research. This case serves as another compelling example of the heterogeneous presentation of LGI1 autoimmune encephalitis, where it should be considered in older adults presenting with new focal onset seizure, especially atypical or stimulus-sensitive, avoiding misdiagnosis with neuromyotonia or CIDP. A common theme, however, is the rapid improvement in symptoms once an effective treatment is initiated early in the disease course.
Supplemental Material
Footnotes
Consent to Participate
Informed consent has been obtained from the patient for submission of this case report.
Author Contributions
Ashutosh Gupta, Cooper university hospital, Camden, NJ, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data. Miles D. Holmes, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data. Divyanshu Dubey, Mayo clinic, Rochester, MN. Drafting the manuscript, data acquisition, study design, interpretation of data. Shirin Jamal Omidi, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data. Megan Goyal, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data. Shitiz Sriwastava, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data. Rajesh K. Gupta, University of Texas Health science center, Houston, Texas. Drafting the manuscript, data acquisition, study design, interpretation of data.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.