
Abstract
Immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome is a rare autosomal recessive disorder characterized by immunodeficiency, centromeric instability, and facial dysmorphism. We report four Chinese pediatric patients with ICF syndrome diagnosed between 2018 and 2021. All patients presented with recurrent infections due to hypogammaglobulinemia. Whole-exome sequencing identified four novel mutations: CDCA7 c.695delC (p. Val233Ter), HELLS c.560T>G (p.L187W), ZBTB24 compound heterozygous (c.1228_1229delinsC and c.649_652del), and ZBTB24 homozygous (c.1208A>G, p.H403R). To our knowledge, this is the first report of CDCA7 p. Val233Ter and HELLS p.L187W mutations globally. These findings expand the mutational spectrum of ICF syndrome.
Introduction
Immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome is a rare autosomal recessive disorder and one of the few known genetic diseases involving constitutive abnormalities in genomic methylation patterns. Other conditions include multilocus imprinting disturbances caused by mutations in ZFP57, ZNF445, or SCMC components (Xu et al., 1999). Most patients succumb to infections before adulthood due to reduced serum immunoglobulin levels. Since its first description in 1979, only approximately 120 cases have been reported worldwide (Weemaes et al., 2013). In China, only five cases have been publicly reported to date (Yang et al., 2021; Lu et al., 2019; Hu et al., 2019). Based on causative genes, ICF syndrome is classified into four subtypes: ICF1 (DNMT3B), ICF2 (ZBTB24), ICF3 (CDCA7), and ICF4 (HELLS; Thijssen et al., 2015). This report presents four Chinese pediatric patients with ICF syndrome, including novel mutations in CDCA7 and HELLS. Recently, compound heterozygous mutations in UHRF1 were also reported in an atypical ICF case (Unoki et al., 2023).
Case Report
Whole-exome sequencing was performed on peripheral blood DNA from the four patients and their parents. Sanger sequencing validated the identified variants. Pathogenicity was assessed according to ACMG guidelines. The following reference sequences were used: ZBTB24 (NM_014797.2; NP_055612.2), HELLS (NM_018063.4; NP_060533.2), and CDCA7 (NM_018958.3; NP_665809.1). The study was approved by the Ethics Committee of Kunming Children’s Hospital (Approval No. 20121-03-72-324-k01). Written informed consent was obtained from all guardians.
Case 1: A 6-year-old Yi boy presented with recurrent fever, cough, and infections since 7 months of age, including suppurative otitis media and skin pustules. He had short stature but normal development. Parents were consanguineous. Laboratory findings showed decreased IgG and IgM. EEG revealed bilateral frontal slow waves; brain MRI showed widened sulci and right thalamic signal abnormality. Karyotype analysis showed 46, XY, t(1;16)(q12; q11). Multiradial configurations involving chromosomes 1, 9, and 16 were observed, consistent with the centromeric instability hallmark of ICF syndrome. Genetic testing identified a homozygous HELLS c.560T>G (p.L187W) variant. He receives regular IVIG therapy.
Case 2: A 9-year-old Han Chinese boy presented with recurrent upper respiratory tract infections and developmental delay. Since 6 months of age, he had multiple hospitalizations for pneumonia with pulmonary consolidation. He had skeletal deformities (pectus carinatum and bowlegs), distinctive facial features (blepharoptosis, low nasal bridge, and broad mandible), and intellectual delay. The parents were non-consanguineous. Immunological evaluation showed decreased IgG and IgM. Karyotype analysis was normal, and no multiradial configurations involving chromosomes 1, 9, or 16 were detected. Genetic testing identified a homozygous ZBTB24 c.1208A>G (p.H403R) variant (VUS). This variant has not been previously reported in public databases (ClinVar, gnomAD) and is therefore considered novel. His parents were non-consanguineous and both were heterozygous carriers. He continues regular IVIG therapy.
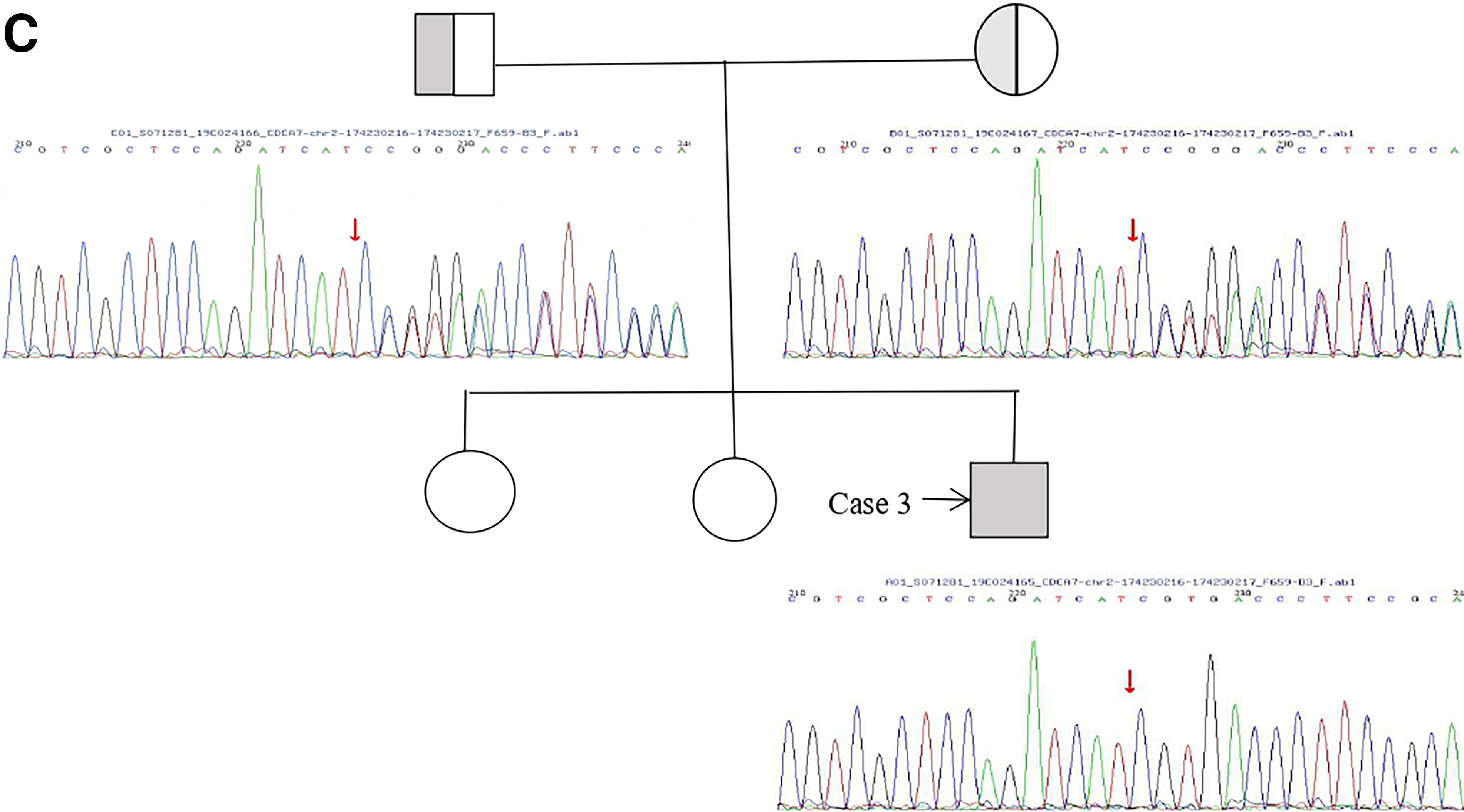
Case 3: A 5-year-1-month-old Hui boy had recurrent pneumonia since infancy. He had pectus carinatum and bowlegs but no obvious facial dysmorphism. Growth and motor development were delayed. The parents were non-consanguineous. Immunology showed decreased IgG, IgM, IgA, reduced CD4+ T cells, and elevated CD19+ B cells. Chest CT showed lung consolidation, bronchiectasis, and empyema. Karyotype was normal, and no multiradial configurations involving chromosomes 1, 9, or 16 were detected. Genetic testing identified a homozygous frameshift variant in CDCA7. The variant corresponds to the major CDCA7 transcript (NM_018958.3; NP_665809.1), in which the nucleotide position is c.695delC resulting in p. Val233Ter. The original laboratory report used an alternative transcript annotation (c.932delC, p. Ser311fs). He received IVIG and anti-infective therapy but still had mild recurrent infections.
Case 4: A 1-year-old Han Chinese boy presented with recurrent pneumonia since 4 months of age. Facial abnormalities included hypertelorism, low-set ears, and left esotropia. He also had intellectual and motor developmental delay, malnutrition, and rib flaring. The parents were non-consanguineous. Karyotype analysis was normal, and no multiradial configurations involving chromosomes 1, 9, or 16 were detected. Genetic testing identified compound heterozygous mutations in ZBTB24: c.1228_1229delinsC (p. Pro410AspfsTer11) (novel) and c.649_652del (p. Glu217LysfsTer90) (previously reported). He currently receives regular IVIG therapy.
The pedigrees and Sanger sequencing validation of the four families are summarized in Figure 1. The locations of the identified mutations on the corresponding protein domains are shown in Figure 2. The clinical and genetic characteristics of all patients are listed in Table 1, and their immunological phenotypes are summarized in Table 2.
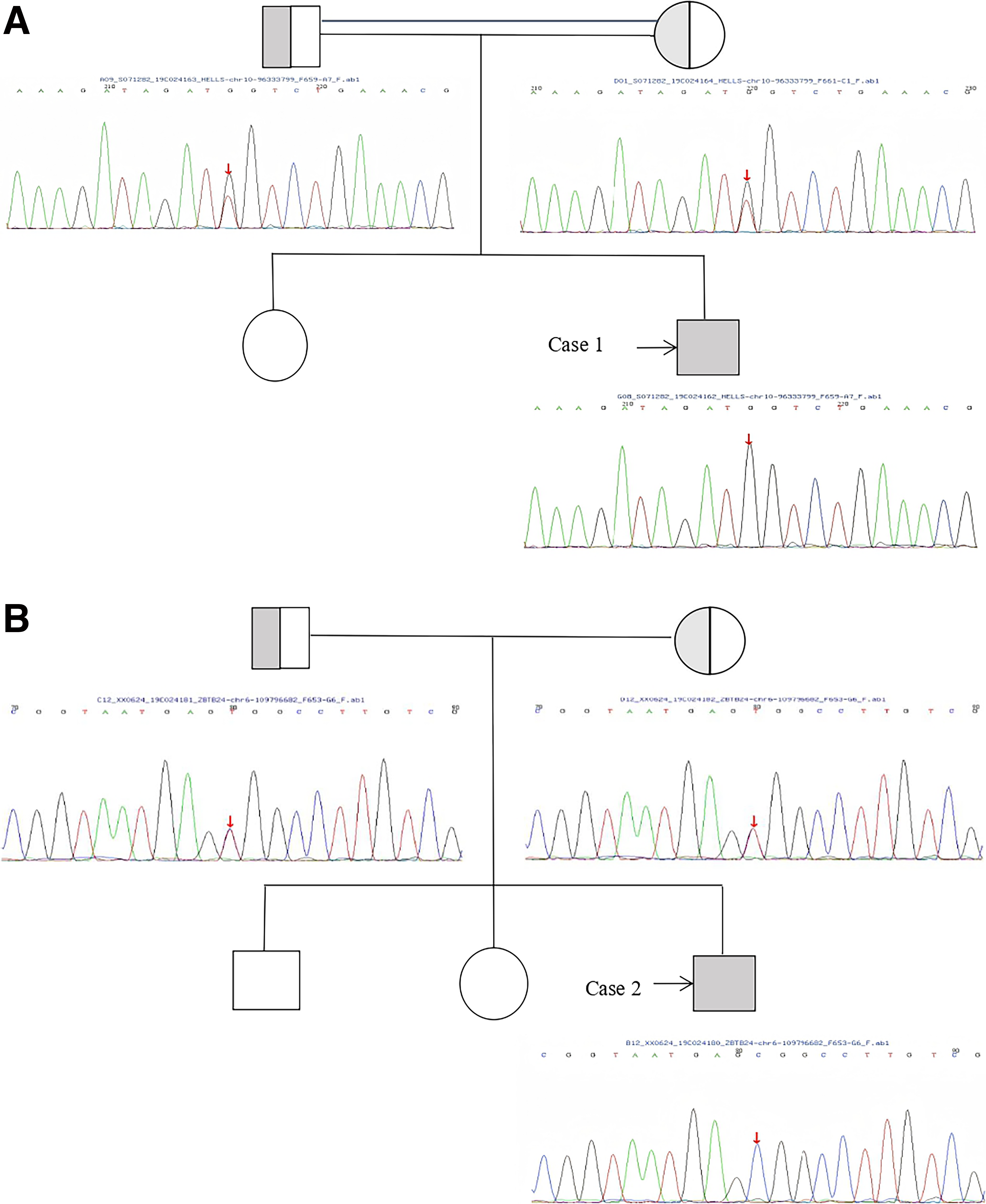
Pedigree analysis and Sanger sequencing validation of the four ICF syndrome families.
Continued
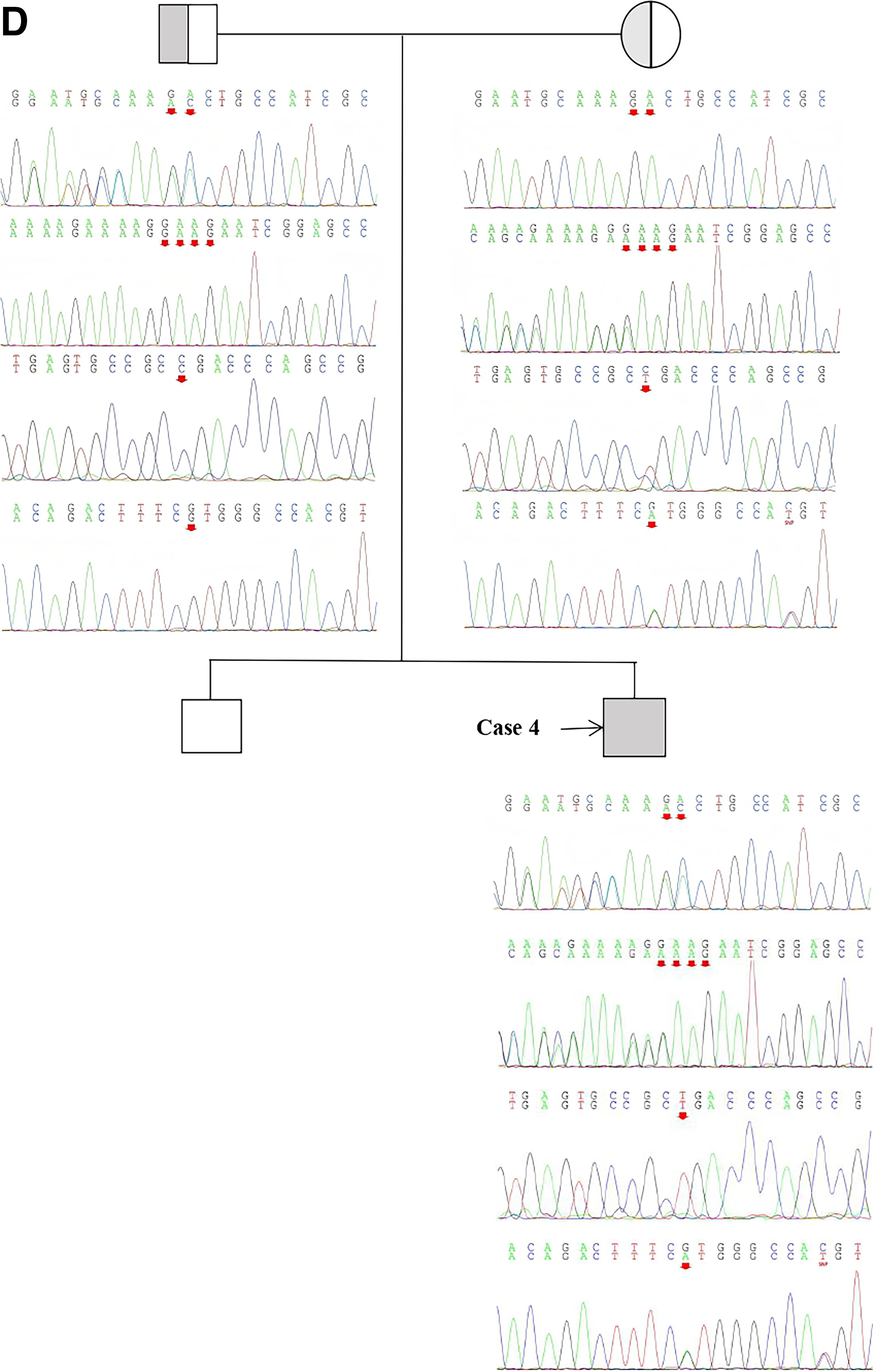
Continued
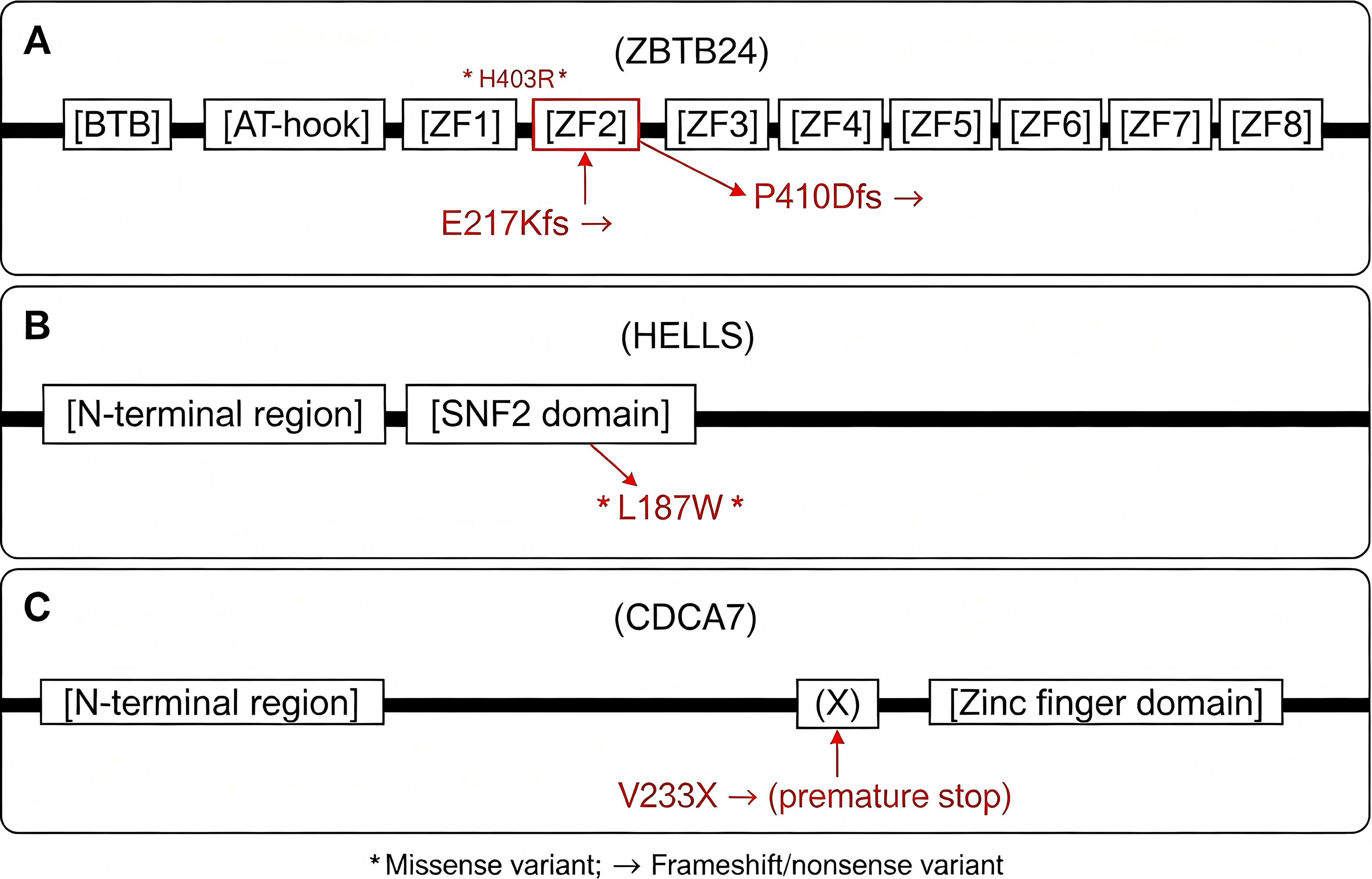
Schematic diagram of protein domains and mutation locations.
Demographic and Genetic Characteristics of the Cohort
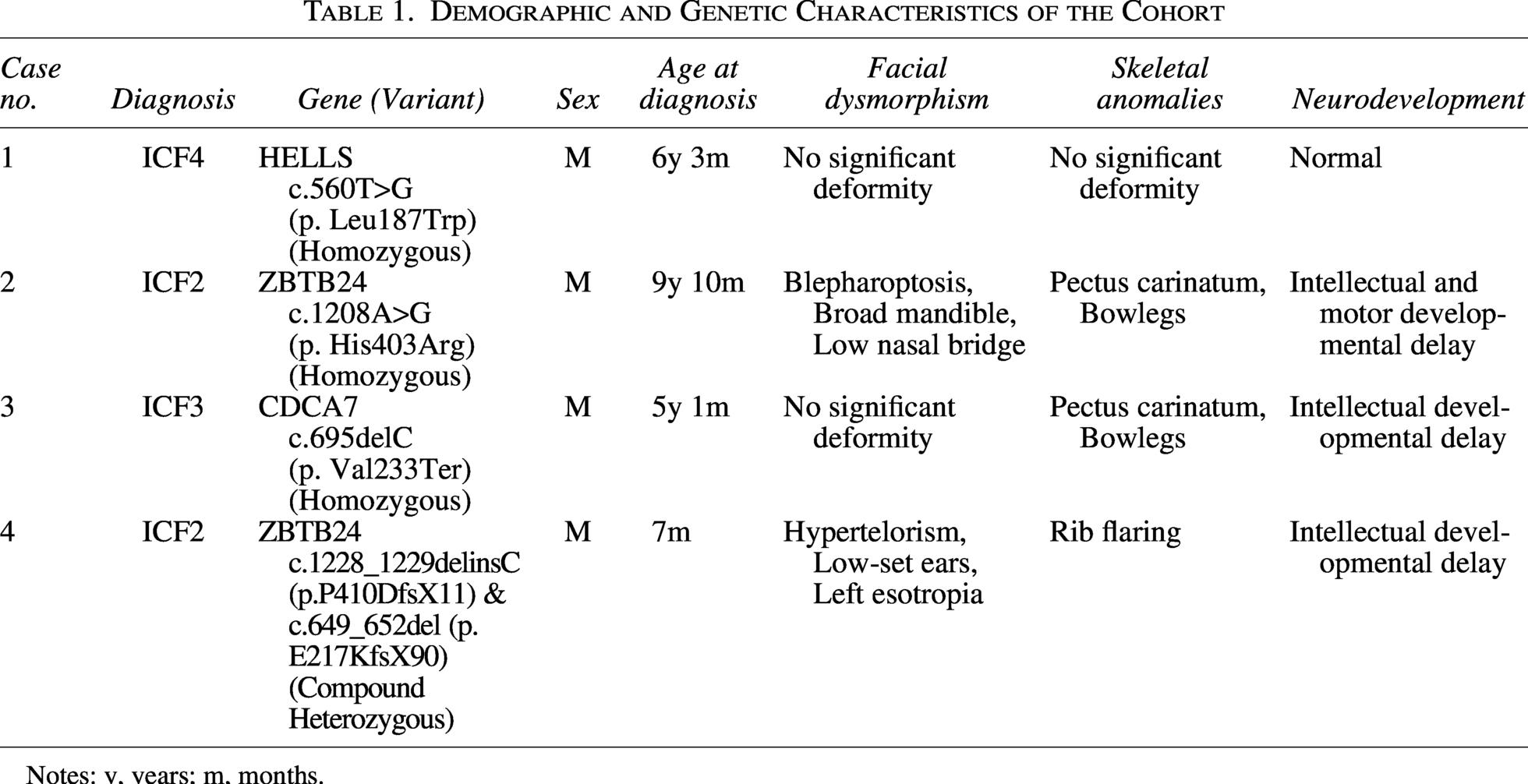
Notes: y, years; m, months.
Summary of Immunological Phenotypes

Note: ↓, decreased; N, normal; ↑, elevated. Ig levels and lymphocyte subsets are interpreted against age-matched normal ranges.
Discussion
We identified potential pathogenic variants in ICF-associated genes (ZBTB24, CDCA7, and HELLS) in four Chinese pediatric patients. Notably, these include two first-reported variants (CDCA7 p. Val233Ter and HELLS p.L187W) and one novel ZBTB24 variant (c.1228_1229delinsC). The primary reason for medical attention in all patients was recurrent infections secondary to hypogammaglobulinemia, the most prominent clinical feature of ICF syndrome (Nitta et al., 2013).
In Case 1, the HELLS variant (c.560T>G, p.L187W) has not been previously reported. REVEL predicted this variant as deleterious. Parental consanguinity and heterozygous carrier status support an autosomal recessive inheritance pattern. Case 3 carries a homozygous frameshift mutation in CDCA7 (p. Val233Ter). Despite the absence of significant facial dysmorphism, the patient presented with recurrent pneumonia, B-cell functional deficiency, and low IgG and IgA, consistent with the immunological profile of ICF3 (Jenness et al., 2018).
To assess the potential pathogenicity of the two novel missense variants (HELLS p. Leu187Trp and ZBTB24 p. His403Arg), we performed in silico prediction using PolyPhen-2. HELLS p. Leu187Trp was predicted to be probably damaging (score 1.000). In contrast, ZBTB24 p. His403Arg was predicted to be benign (score 0.000). As the reviewer noted, this benign prediction is not unexpected because the variant, although located within the fifth zinc-finger domain, does not disrupt the critical C2H2 motif. Of note, PolyPhen-2 uses UniProt O43167 (the longer ZBTB24 isoform of 517 amino acids) as the reference, and the histidine at position 403 is predicted to be non-essential for the C2H2 structure. Despite the benign in silico prediction, the homozygous inheritance of this variant in a patient with typical ICF2 clinical features (recurrent infections, hypogammaglobulinemia, skeletal anomalies, and developmental delay) suggests that it may still contribute to the disease, possibly through a less severe or hypomorphic mechanism. Functional studies are warranted to confirm its pathogenicity. The locations of all identified variants within protein domains are illustrated in Figure 2.
ZBTB24 encodes a transcriptional regulator linked to DNA methylation regulation and immune system development (Ren et al., 2019). Case 2 had a homozygous ZBTB24 variant (p.H403R) currently classified as VUS. Case 4 carried two ZBTB24 frameshift mutations (c.1228_1229delinsC and c.649_652del), both likely leading to protein truncation and loss of function (Nie et al., 2022). In addition, we identified a novel ZBTB24 frameshift variant c.1228_1229delinsC (p. Pro410AspfsTer11) in Case 4, which expands the mutational spectrum of ZBTB24.
This study has limitations, including a small sample size from a single center and lack of functional validation (e.g., methylation analysis or protein expression studies). Functional studies are needed to confirm the pathogenicity of these novel variants.
Conclusion
This report expands the mutational spectrum of ICF syndrome in the Chinese population with novel CDCA7, HELLS, and ZBTB24 variants. Functional studies and multi-center collaboration are needed to further confirm their pathogenicity.
Ethics Approval and Consent
This study was approved by the Ethics Committee of Kunming Children’s Hospital (No. 20121-03-72-324-k01). Written informed consent was obtained from the legal guardians of all patients.
Footnotes
Acknowledgments
The authors are grateful to all data collectors. Y.W. helped with the preparation of figures for this article.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
The work was supported by the