
Abstract
Introduction:
Long-term preservation of biological material for biomedical or animal conservation purposes currently relies on ultracold temperatures. However, almost complete dehydration of cells in the presence of trehalose could allow cost-effective and flexible storage at ambient temperatures.
Objective:
The study aimed to characterize optimal trehalose-loading methods before passive air-drying and explore different storage options using domestic cat primary fibroblasts as a model.
Methods:
In Experiment 1, cells were loaded with trehalose using optimized electroporation (EP), thermal shock, or cold-responsive nanoparticles (CRNPs) containing trehalose. An evaluation of the cell viability after freezing and thawing was first conducted to compare trehalose protections conveyed by the different loading methods. In Experiment 2a, cells loaded with trehalose were passively air-dried for 2 days (once dry state was confirmed) and then assessed for viability, DNA integrity, or proliferative ability. In Experiment 2b, dried cells were stored for up to 4 weeks either at ambient temperatures, ambient temperatures under vacuum, 4°C, or −20°C while being assessed for viability every 24 hours for the first 5 days of storage and at the 2- or 4-week timepoint.
Results:
Loading cells with trehalose using EP and thermal shock methods achieved cryoprotective levels, but CRNPs did not enable them to reach that. After reaching the dry state, cells from all trehalose-loading methods and untreated controls had viable cells. Additionally, trehalose-loaded cells from all methods had no increase in DNA damage after drying when compared with fresh controls. Following each method, a small number of cells retained proliferative ability. Storage at 4°C helped prolong viability through the initial 5 days compared with other storage conditions; however, no cells were viable after 2 weeks.
Conclusion:
Cells maintain viability, DNA integrity, and proliferative ability after reaching the dry state; however, storage conditions beyond 5 days must be improved.
Introduction
Biobanking involves the preservation of biological materials, including living animal cells, for long-term at freezing temperatures. Current biobanks rely on ultracold freezers (−80°C) or storage at −196°C in tanks filled with liquid nitrogen. It is a fundamental tool for understanding and sustaining genetic diversity from endangered animal species and, ultimately, for safeguarding global biodiversity. 1 Importantly, these frozen samples can also be used to produce live offspring via assisted reproduction and rescue species from the brink of extinction. Biobanks are also critically important to biomedical science, where research is dependent on large numbers of high-quality samples.2,3 With increasing threats to biodiversity and growing needs to bank samples for the future, it is critical to (1) store high-quality samples (including cells that can reanimate and function normally after storage) and (2) reduce the reliance on electricity or liquid nitrogen for long-term storage. 4 More strategies therefore are urgently needed to easily preserve and store genetic materials and to decrease the maintenance costs and carbon footprint of biobanks.
Somatic cells, such as fibroblasts, can be conveniently obtained from animals in a wide variety of tissue biopsies and then multiplied in vitro. Once banked, somatic cells can also be used for various research fields from evolutionary genetics to stem cell technologies. 1 Thus, biobanking of somatic cells offers more options to understand and sustain genetic diversity than DNA samples. Additionally, biobanking cell lines is critical to biomedical research because it is a way to preserve high quality and standardized biological material which ensures consistency across long-term studies, and patient-derived cell lines that facilitate the development of personalized medicine. 2 Cell lines also allow for disease modeling and the study of cellular behavior which are crucial to drug development and therapeutic interventions. 5
Over the last decade, our laboratory has developed drying techniques as an alternative to traditional cryopreservation methods. Mimicking the protective mechanisms discovered in drought-resilient species, this approach utilizes trehalose (a nonreducing disaccharide sugar synthesized by these organisms) to protect cells from dehydration damage by maintaining the three-dimensional conformation of molecules, preventing collapse or aggregation of cellular structures, and stabilizing cellular components in the amorphous trehalose glass under dried conditions. 6 Previous studies have demonstrated that intracellular delivery of trehalose by cold-responsive nanoparticles (CRNPs) enhances dehydration tolerance of oocytes. 7 Trehalose-loaded samples can potentially be safely dehydrated to eventually allow for storage at less energy-demanding temperatures, with ambient temperatures being the ideal.8–10 While encouraging results have been reported for gametes, more research is still needed to apply this dry-preservation strategy to somatic cells, including fibroblasts. There are some reports of using passive-air drying to allow human mesenchymal stem cells to survive being shipped overnight across the United States without culture medium at ambient temperatures.11,12 While this short-term retention of viability could be helpful for transporting samples, the benefits of long-term storage in the dry state are even larger. Freeze drying is commonly used to preserve protein drugs and vaccines; however, there are no reports of being able to use this technique on whole cells, and it also would require specialized equipment.13,14 We aimed to utilize passive-air drying, building on initial reports of mesenchymal stem cells surviving short periods in a dry state.11,12,15 This reduces the need for additional specialized equipment, which can be difficult to procure in fields that are less funded than biomedical sciences, such as wildlife conservation, which can also take place in remote locations. For this study, we focused on using primary fibroblasts derived from domestic cats, a valuable biomedical model and also a model for wild and endangered felids. Since the ultimate goal of dry storage is to preserve a valuable genetic resource for future use, we not only assessed the viability of these cells, but also their DNA integrity to ensure the genetic information was still intact, and their proliferative ability to ensure that the cells could continue self-sustaining for at least several passages.
The overall objective of the present study was to characterize the optimal trehalose-loading methods before passive air-drying and storage of fibroblasts and to evaluate cellular response to drying and rehydration by assessing cell viability, DNA integrity, and proliferative ability. The main goal was to advance the development of new preservation strategies to sustainably store somatic cells for biomedical purposes or to ensure genetically diverse populations of wild species.
Methods
Animals and reagents
All reagents were from Sigma unless otherwise noted. Experiments described did not require the approval of the Animal Care and Use Committee of
Fibroblast culture
Sharp dissection was used to isolate the oviduct from the rest of the reproductive tract and rinsed in PBS. A small piece of oviduct ∼1 cm in length was placed into a 500 µL drop of 2 × antibiotic fibroblast medium (DMEM/F12 (Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco), 1× GlutaMAX (Fisher), 1× MEM nonessential amino acid solution, 180–210 IU/mL penicillin, 250 μg/mL streptomycin, and 20 μg/mL gentamicin) and minced with a scalpel blade. The minced tissue was transferred to a fresh 60-mm culture dish, and 5 mL of 2× antibiotic fibroblast medium was added. The dish was cultured in a 38.5°C incubator with 5% CO2 for 48 hours to allow fibroblasts to grow out of the tissue pieces and attach to the bottom of the dish (passage 0). Then the tissue pieces were removed, and the medium was exchanged. Culture continued with 2 × antibiotic medium replaced at least twice per week until the dish reached 90%–95% confluency. To passage cells, the dish was rinsed once with PBS, no calcium, no magnesium (PBS -Mg -Ca, Gibco), and then cells were dissociated with 1 mL TrypLE for 5–10 minutes until all cells were detached from the dish. The TrypLE was inactivated with 1 mL DMEM/F12, and cells were pelleted at 500 × g for 5 minutes in a 2-mL Eppendorf tube. Cells were suspended in Complete Fibroblast Medium (DMEM/F12 supplemented with 10% FBS), 1× GlutaMAX (Fisher), 1× MEM nonessential amino acid solution, (90–105 IU/mL penicillin, and 125 μg/mL streptomycin) and plated on 35-mm dishes (Passage 1). Cells were incubated as above until dishes reached confluency of at least 90% and then were used for experiments. All cells used for experiments were passage 1.
Experimental design
A graphical overview of the experimental design for this study is summarized in Figure 1. In Experiment 1, we first refined the methods of electroporation by assessing cell viability in response to 27 different EP methods and then further testing the loading efficiency of the methods that led to the optimal viability of 50%–60% by using a fluorescent proxy for trehalose (at least three replicates involving cells from different cats each time). This allowed us to determine the EP method that was able to load the maximum amount of trehalose while preserving 50%–60% viable cells to go into the drying process. Cells were also loaded with trehalose by using a thermal shock method and using CRNPs. Viability after cryopreservation was used as a control to determine whether cryoprotective levels of trehalose were loaded into the cells using these methods. For all assays in this study, at least three replicates for each method were performed, each using cells derived from a different cat, and at least 100 cells per replicate were assessed. In Experiment 2, cells were dried using a passive air-drying method and were immediately rehydrated after the 2-day drying period and assessed for viability, DNA integrity, and proliferative ability, or they were stored at ambient temperature, ambient temperature under vacuum, 4°C, or −20°C and then rehydrated and assessed for viability. Experiments were repeated at least three times with cell lines from different cats.
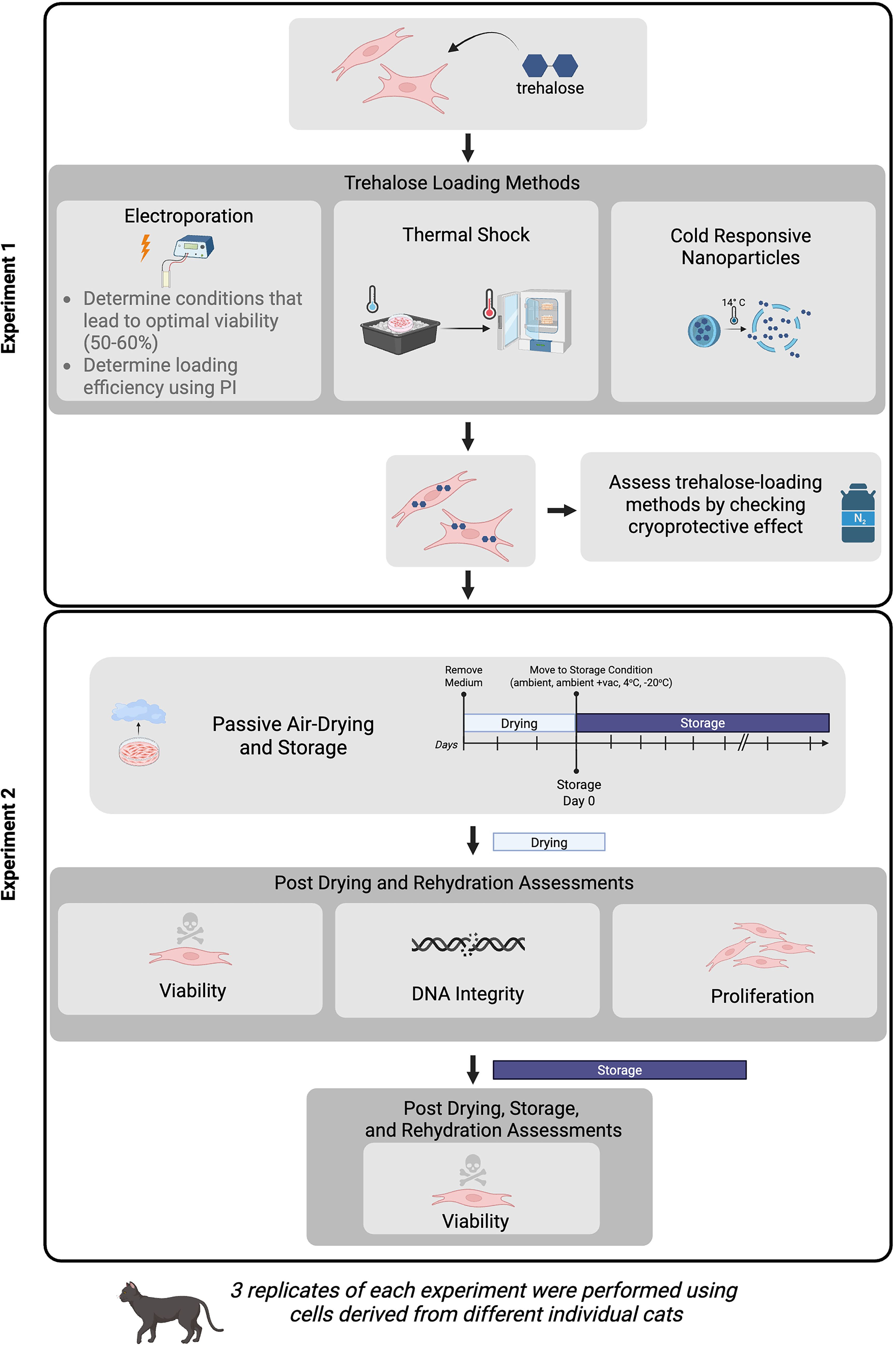
Experimental design showing methods used in Experiment 1 to load primary fibroblasts with trehalose and assess them using cryopreservation, and the drying and storage timeline in Experiment 2 and the assessments performed after drying and immediate rehydration or drying, storage, and rehydration. Created in BioRender. Sosnicki (2026) https://BioRender.com/2dna0mk.
Electroporation and cell loading assessment
Cells were dissociated from the culture dish using TrypLE as above and resuspended in 200 µL electroporation buffer (EP buffer) containing 0.3 M trehalose in DMEM/F12. The concentration of trehalose was determined based on studies that determined osmotic stress was not detrimental to cellular health at a very similar concentration of 290 mM in mouse myeloma cells loaded by electroporation. 16 This suspension was added to 0.2 cm Gene Pulser Xcell electroporation cuvettes (Bio-Rad) and electroporated using a square wave protocol on a Gene Pulser Xcell (Bio-Rad). Combinations of pulse strength (120, 140, or 160 V), pulse length (15, 20, 25 ms), and number of pulses (1, 2, or 3 pulses with a pulse interval time of 10 seconds) were evaluated. Cells in EP buffer were then moved to a 500-µL Eppendorf tube and allowed to recover at 38.5°C for 15 minutes to ensure live cells had complete membrane closure before adding the trypan blue dye to assess viability (see below). Optimal EP method for transfection should result in the highest gene expression activity paired with the highest cell viability, with the most optimized conditions for many cell types having a viability of ∼50%.17,18 For this study, we therefore defined optimal EP method to yield a 50%–60% viability after electroporation, which strikes the balance between forming large enough holes in the membranes to load the cells with the maximum amount of trehalose while maintaining the most viable cells to go into the drying process. To determine the loading efficiency of the EP methods that yielded viability within this range, we adapted a method that was previously reported using a different cell type 16 in which propidium iodide (PI), a similarly sized, fluorescent proxy for trehalose, was added to the EP buffer at a concentration of 20 µg/mL. After EP, cells were allowed to recover for 15 minutes at 38.5°C and then the cells were costained with calcein AM to identify live cells. After capturing images of the cells, we measured the PI fluorescence intensity in only the live cells by creating a mask in ImageJ using the calcein AM staining. See Figure 2A for representative images of calcein AM and PI staining as well as the regions of interest that were determined from the calcein AM staining.
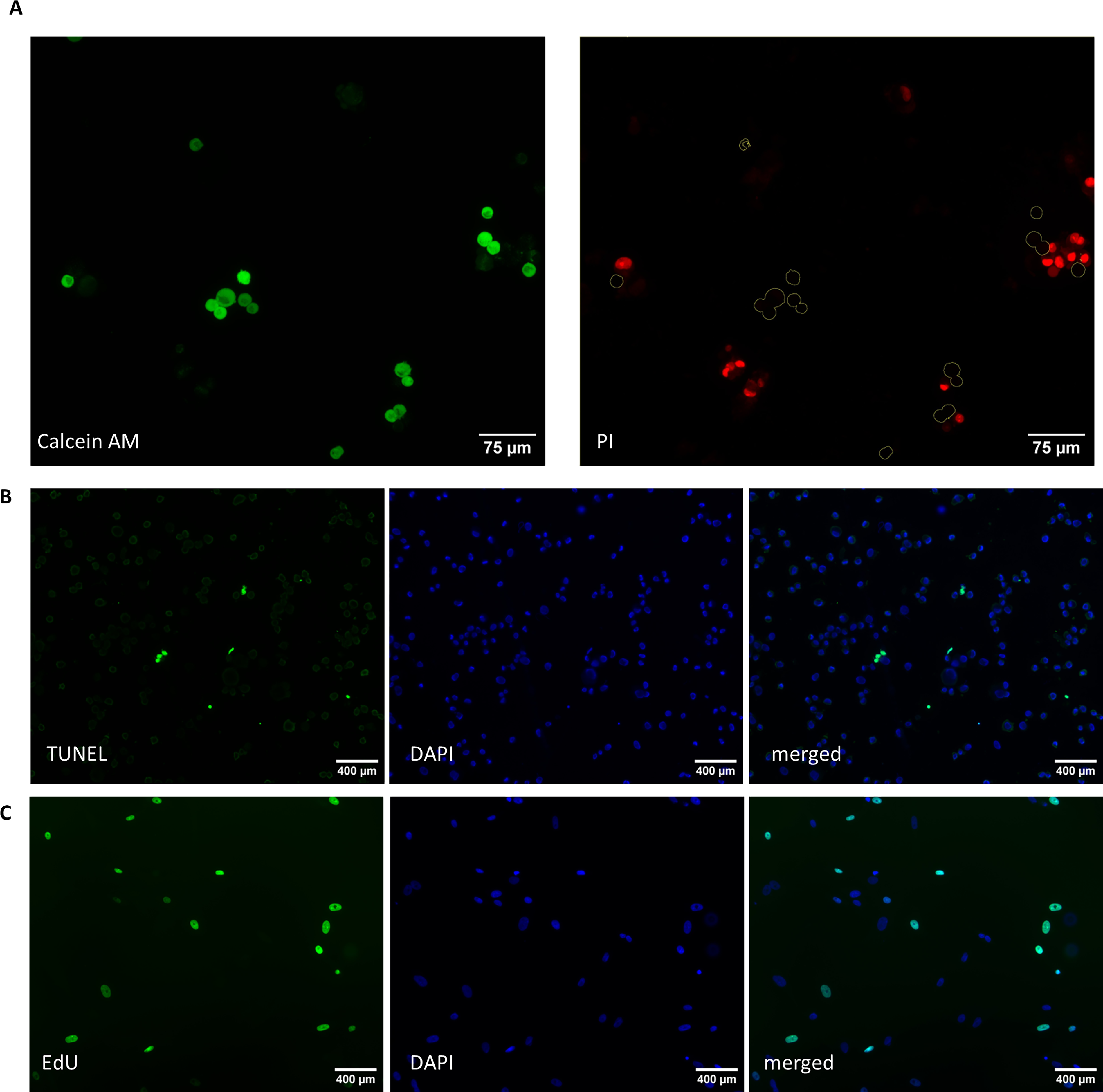
Representative images of
Cell loading through thermal shock
The thermal shock protocol was adapted from a previously published report that found this method conferred desiccation tolerance to human foreskin fibroblast cell lines. 12 Culture medium was removed and replaced with 50 mM trehalose and 3% glycerol in DMEM/F12. Dishes were immediately placed on ice for 5 minutes and then transferred back to the incubator at 38.5°C for 10 minutes. This protocol mimics others, such as bacterial heat transformation that utilize rapid temperature changes causing rapid phase transitions in the lipid bilayer of the cell membrane, which in turn can increase membrane fluidity and cell permeability.
Cold-responsive nanoparticle synthesis, characterization, and cell loading
CRNPs were synthesized using a double emulsion (water-in-oil-in-water) method adapted from a previously reported protocol. 4 Briefly, a solution of 1.3M trehalose in ultrapure water was prepared as the first water phase. The oil phase consisted of 15 mg of PLGA (poly D, L-lactide-co-glycolide acid terminated), 30 mg of pNIPAM-B (poly N-isopropylacrylamide-cobutylacrylate), and 10 mg of PF127 (Pluronic F-127) dissolved in 2 mL of dichloromethane (DCM). To form the first emulsion, 300 µL of the 1.3 M trehalose solution was added to the oil phase and emulsified for 1 minute at 84% amplitude using a FB120 sonicator with a maximum voltage of 120 V (Fisher Scientific). This first emulsion was then added dropwise to 10 mL of 2% polyvinyl alcohol solution and emulsified for 1 minute at 100% amplitude to obtain the double emulsion. The nanoparticle suspension was then stirred on a 37°C stir plate overnight to evaporate the DCM.
The CRNPs were assessed for morphology using transmission electron microscopy (TEM). Briefly, the nanoparticles were resuspended in deionized water and added to the 200 mesh copper grids with pure carbon film (Ted Pella, Redding, CA) before (Supplementary Fig. S1A) or after cold treatment (15 minutes in 4°C refrigerator; Supplementary Fig. S1B). The samples were stained with uranyl acetate solution (2%, w/w) and imaged by a JEOL (Akishima, Tokyo, Japan) JEM 2100 LaB6 TEM. The size of nanoparticles (Supplementary Fig. S1C) was assessed using dynamic light scattering by dispersing 0.5 mg of nanoparticles in 2.5 mL of deionized water at ambient temperature.
Cells were loaded with CRNPs by first collecting the nanoparticles by centrifugation of 2 mL of nanoparticle suspension at 9600 × g for 10 minutes at ambient temperature and resuspending in 2 mL CRNP medium (DMEM/F12 supplemented with 1× GlutaMAX, 1× MEM nonessential amino acid solution, 90–105 IU/mL penicillin, and 125 μg/mL streptomycin). Cells were loaded with CRNPs by adding 2 mL of CRNPs in CRNP medium to each 35-mm dish of cells to be treated and incubated at 38.5°C for 4 hours. After incubation, CRNP medium was removed, and cells were rinsed once with PBS and then 1 mL of PBS was added to the dish, and the cells were cold-treated at 14°C for 15 minutes to release the trehalose from the intracellular CRNPs. This cell loading method and cold treatment were confirmed to deliver CRNP cargo on feline primary fibroblasts using nanoparticles containing fluorescent doxorubicin (DOX) and then examining the cells using fluorescent microscopy (Supplementary Fig. S1C). This method using the fluorescent properties of DOX to track cellular uptake and release of CRNPs has previously been validated.4,7
Passive air-drying, storage conditions, and rehydration
Culture medium was removed from 35-mm dishes containing adherent cells using vacuum aspiration, being sure to remove as much of the liquid as possible to allow for more uniform drying times. While it would be ideal to control ambient temperature and relative humidity to truly standardize this drying process, these parameters were outside of our control. During this study, the ambient temperature ranged from 17.0°C and 22.2°C and the relative humidity ranged from 17.5% to 71.8%. The dish lids were placed back on the dishes, and one layer of parafilm was wrapped around to seal the dish. The dishes were protected from light in a dark cabinet at ambient temperature during the drying process. Cells were left undisturbed during the drying process and then were moved to storage conditions or were immediately used for other assays.
The storage period began after 2 days of drying when the cells had reached the dry state (based on water content measurements reported in the results section). Day 0 of storage was the third day after culture medium was removed, and the storage period did not include the initial drying period (see Fig. 1 for a timeline of the drying and storage periods).
Cells were rehydrated by adding 1 mL of warmed DMEM/F12 to the 35-mm dish the cells were dried on and placing the dish in a 38.5°C incubator with 5% CO2 for 30 minutes before assessing for viability or use for other assays described below.
Cell cryopreservation and thawing
To confirm that enough trehalose was entering the cells using the different trehalose-loading methods described above, cells were treated with each of these, then were suspended in 1 mL of 0.3M trehalose in DMEM/F12, transferred to cryovials, and then placed in a Styrofoam box before being moved to a −80°C freezer for 24 hours to control their cooling rate. After 24 hours, the cryovials were transferred to liquid nitrogen and stored for at least 24 hours before thawing in a 37°C water bath and immediate assessment for cell viability (see below).
Water content measurement
To determine when cells reached the dry state using the passive air-drying method, water content was measured immediately after the culture medium was removed from untreated cells and then every 24 hours for 5 days. No trehalose-loading methods were performed on cells used to determine the water content during the drying process. Water content of the samples was measured by growing fibroblasts on coverslips placed at the bottom of 35-mm culture dishes. Once the cells were confluent, the medium was removed, and the back of the coverslip (without cells) was blotted on a KimWipe to remove residual culture medium. Water content on Day 0 was obtained by immediately adding the coverslip with adhered cells into a V20 Volumetric KF titrator (Mettler Toledo). For other time points, the coverslip was moved to a new, unused 35-mm culture dish, and parafilm was used to seal the lid on to mimic the drying process of cells adhered to the bottom of the dishes for drying experiments. At the desired time point, the coverslip was removed from the dish and immediately placed in the titrator.
Cell viability assessment
Cell viability was assessed by trypan blue exclusion. A cell suspension was mixed 1:1 with trypan blue, and 10 µL was loaded onto a hemacytometer. All cells in the four corner squares of the hemacytometer were counted with distinction between live cells (no trypan blue staining) and dead cells (with trypan blue staining). Viability was calculated as the percentage of total cells that excluded the stain.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick-end labeling) assay after drying
Cells were dried for 2 days, and then the In Situ Cell Death Detection Kit, Fluorescein (Roche) was used per instructions including the use of recommended positive and negative controls to label double-strand DNA breaks and identify cells in the early stages of apoptosis. Three replicates of each method were performed using cells derived from different individuals. Briefly, after drying and rehydration, cells were dissociated from the culture dishes using TrypLE and fixed with 2% PFA for one hour at ambient temperature. Fixed cells were allowed to settle and attach to a charged glass slide (SuperFrost Plus slides). The cells were gently washed with PBS, permeabilized for 2 minutes and washed again with PBS. They were incubated with the reaction cocktail for 1 hour at 37° C. The slides were washed with PBS and then were mounted with VectaShield hardset with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs) and imaged as described below. At least 100 cells across three replicates from each method were evaluated. Cells were manually determined to be positive or negative for terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining, and then the percentage of TUNEL positive cells was calculated using the total number of cells that were stained with DAPI. The EP method after drying and rehydration was excluded after analysis because less than 100 cells were recovered for this assay, likely due to the large number of cells that were killed during the EP process and the reduced number of cells that were intact after drying and rehydration. See Figure 2B for representative images of this assay.
Proliferation assay after drying
Cells were dried for 2 days and then the Click-It EdU Cell Proliferation Kit for Imaging, AlexaFluor 488, was used per instructions, and three replicates of each method were performed, with each using cells derived from different individuals. Briefly, after drying and rehydration, the rehydrated cells were dissociated from the bottom of the culture dish using TrypLE and were plated onto a glass coverslip in a 35-mm dish with 2 mL of Complete Fibroblast Medium containing 5 µM Edu (5-ethynyl-2′-deoxyuridine). Cells were incubated and allowed to attach to the coverslips for 24 hours. The medium was removed, and the cells were fixed, permeabilized, and incubated with the reaction cocktail for 30 minutes at ambient temperature. Slides were washed with PBS and then mounted with VectaShield hardset with DAPI. Cells were imaged as described below and were manually determined to be positive or negative for EdU staining. Very few cells were recovered (<30 per method) and because the assay only included live cells that were able to reattach, it did not include the cells that died during the drying and rehydration process, so statistics were not performed. Instead, this assay indicated that a small subpopulation of cells was able to retain their proliferative ability after reaching the dry state. See Figure 2C for representative images of this assay.
Cell viability assessment after drying and storage under different conditions
Cells were dried, stored, and rehydrated under the conditions described above. After the 2-day drying period, the storage period began. Storage D0 was the assessment at the end of the drying period at ambient temperature before cells were moved to alternate storage conditions (i.e., different temperature or vacuum conditions). Every 24 hours for the first 5 days, and once at the 2-week and 4-week time points, the cells were rehydrated and assessed for viability using the trypan blue assay described above.
Image acquisition and analysis
Cells were imaged using an Olympus BX41 microscope and Zeiss Axiocam 305 mono/color dual camera system. Images were analyzed using ImageJ software, and statistical analyses were performed using GraphPad Prism software (version 6.05). The Shapiro–Wilk normality test was used to determine normal distribution; analysis of variance (ANOVA) was used for data that passed the normality test, and the Kruskal–Wallis test was used for data that was not normally distributed. Tukey’s test was used for multiple comparisons after ANOVA and Dunn’s test was used for multiple comparisons after Kruskal–Wallis.
Results
Effect of different electroporation protocols on cell viability and prediction of the most efficient trehalose-loading protocol
Based on the percentages of viable cells after treatments, a total of 27 combinations of pulse strength (120, 140, 160 V), pulse length (15, 20, 25 ms), and number of pulses (1, 2, 3 pulses; Fig. 3A) were evaluated. Among those combinations, 10 EP methods fell within an acceptable viability range of 50%–60% while untreated controls averaged 79% viable cells (Fig. 3A).
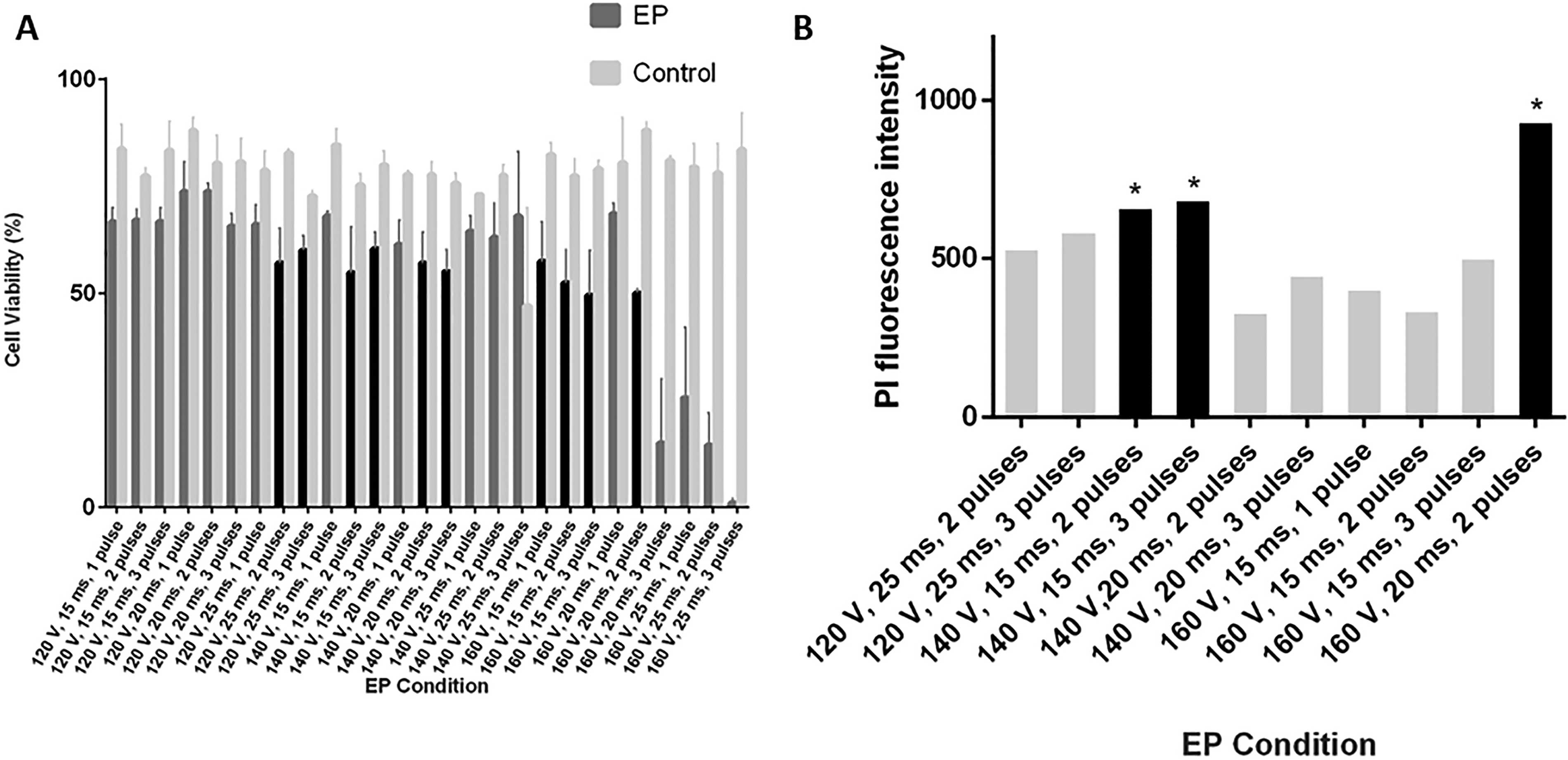
Electroporation optimization methods showing
Those 10 methods were further explored to predict the amount of trehalose that could be loaded into the cells, using PI as a similarly sized, fluorescent proxy for trehalose. Three methods led to the highest PI intensity (140 V, 15 ms, 2 pulses; 140 V, 15 ms, 3 pulses; and 160 V, 20 ms, 2 pulses) that were different from other methods (p < 0.01) but not from each other (p > 0.05, Fig. 3B). One of these methods, 160 V, 20 ms, 2 pulses, was chosen as the optimal EP method to use as a treatment group in subsequent experiments.
Effect of different trehalose-loading methods on cell resilience to cryoprotection
To ensure that enough trehalose was entering the cells using different trehalose-loading methods (optimal EP, thermal shock, or incubation with CRNPs), cells were assessed for viability after standard cryopreservation and thawing (Fig. 4). There was a significant difference in the percentage of live cells that were loaded by EP (33.7 ± 4.4%, p < 0.05) and thermal shock (40.3 ± 11.1%, p < 0.01) compared with untreated cells (0.7 ± 0.7%), but no statistical difference was detected between EP and thermal shock (p > 0.05). There was also no statistical difference between untreated cells and cells loaded with CRNPs (21.3 ± 3.8% SEM, p > 0.05).
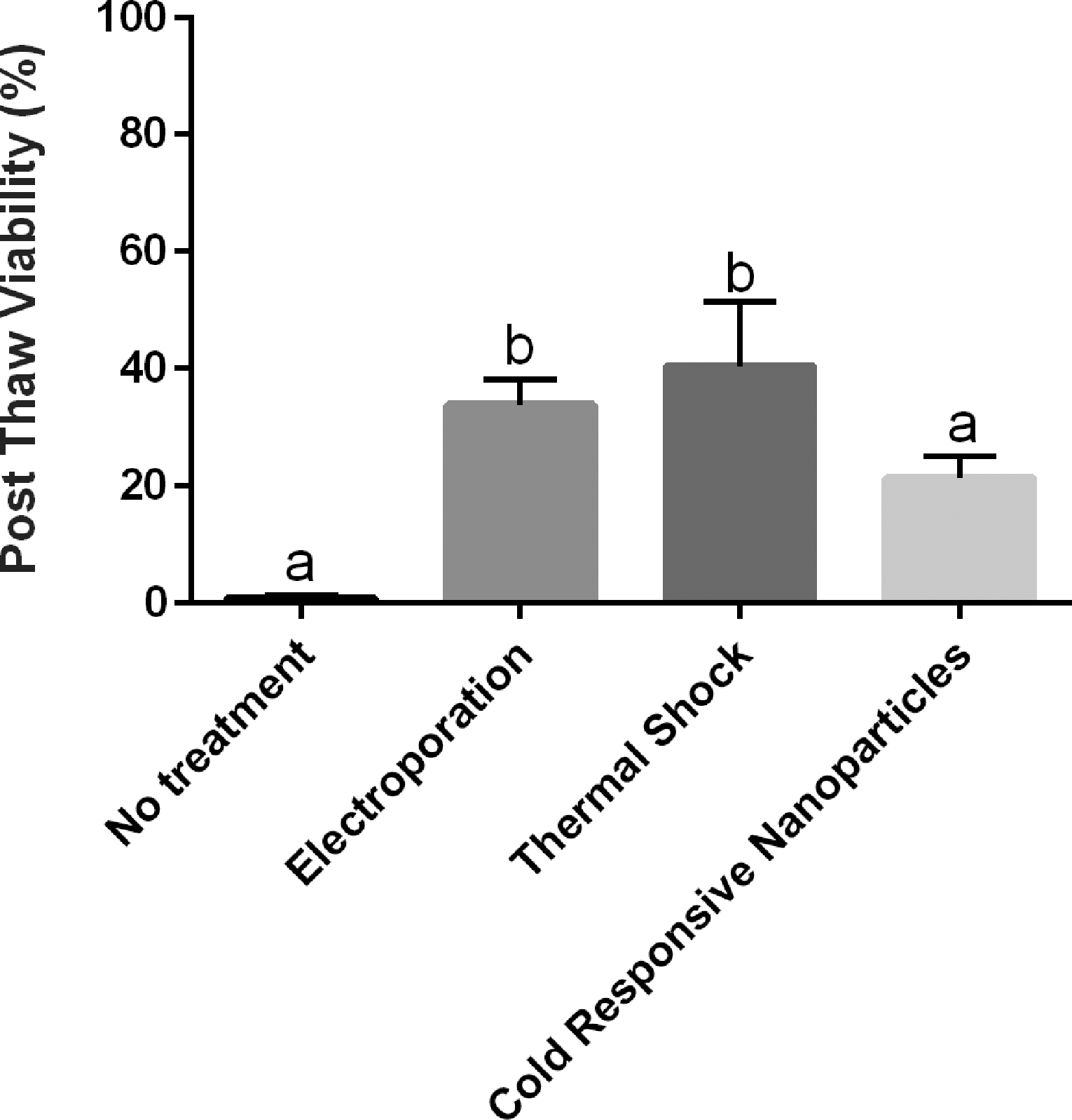
Percentages (mean ± SEM) of viable cells of total cells postthawing following no treatment and different methods to load trehalose via electroporation, thermal shock, or cold-responsive nanoparticles (CRNP). There was no statistical difference between the no treatment and CRNP conditions, but cells treated with electroporation or thermal shock had higher viability postthaw. Different letters above the bars indicate statistical difference (p < 0.05).
Water content of cells during passive air-drying
Using the water content measured on Day 0 as 100%, cells were down to 13 ± 3.8% water by Day 1, 3 ± 0.6% by Day 2, 1.3 ± 0.3% by Day 3 (Fig. 5). After Day 3 of passive drying, minor fluctuations in water content ranged from 1% to 4% (likely due to relative humidity changes in the laboratory). Starting on Day 3 of passive drying, cells reached a stable dry state.
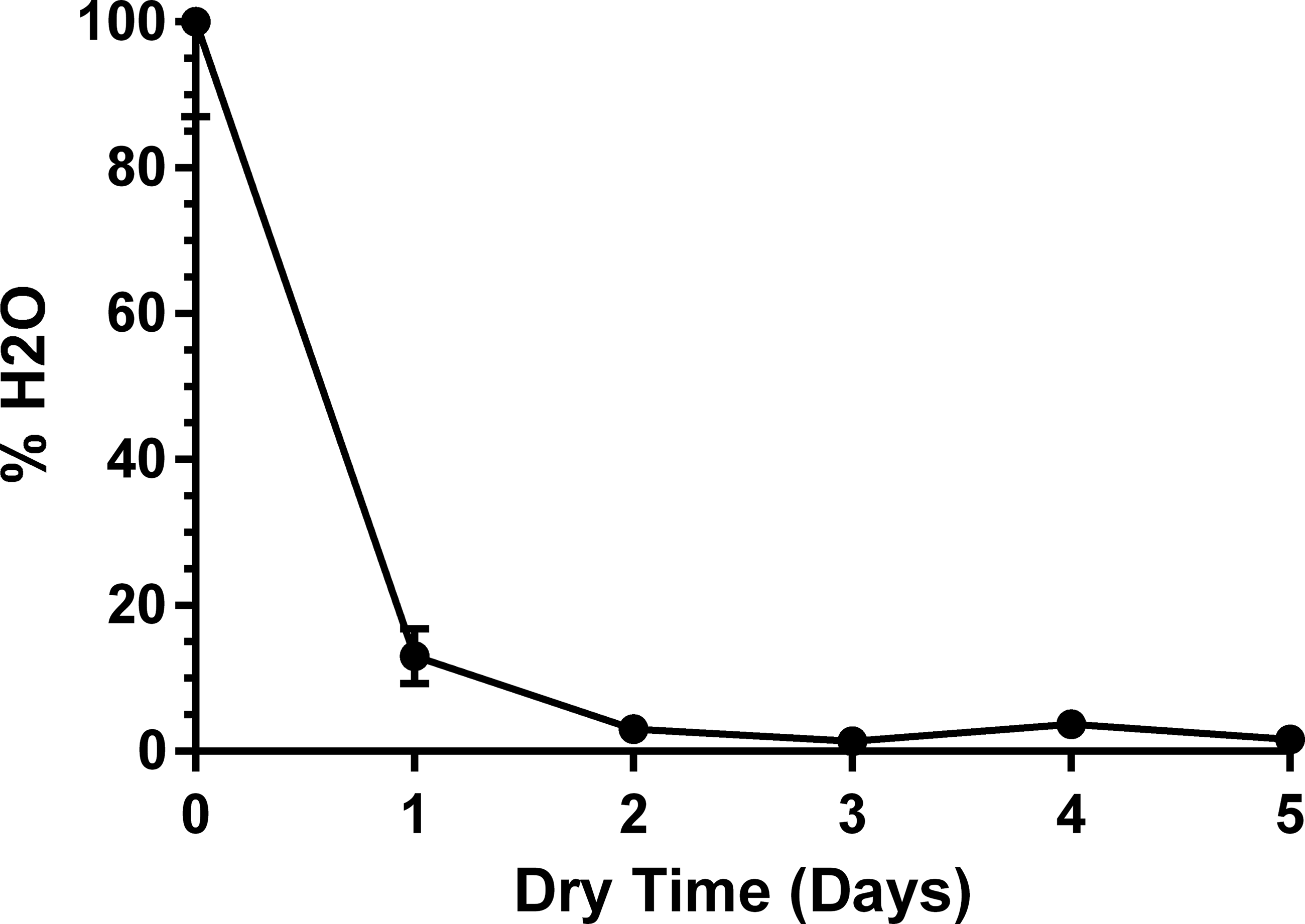
Average water content (mean ± SEM) of cells for the first 5 days after cell culture medium was removed and the drying process had started.
Effect of different trehalose-loading methods on DNA integrity after passive air-drying and rehydration
TUNEL assay was used to assess DNA integrity (by labeling double-strand DNA breaks) of cells after each of the trehalose-loading treatments to first determine the effect of the treatment itself, and after loading and reaching the dry state followed by immediate rehydration (Fig. 6). There was a difference in the percentage of TUNEL positive cells between the control (absence of treatment, 4.2 ± 0.9%) and the absence of treatment followed by drying (28.8 ± 6.4%, p < 0.05), but no difference between any of the other methods (p > 0.05; Fig. 6). The electroporation followed by drying was excluded from analysis due to insufficient numbers of recoverable cells (<100 cells).
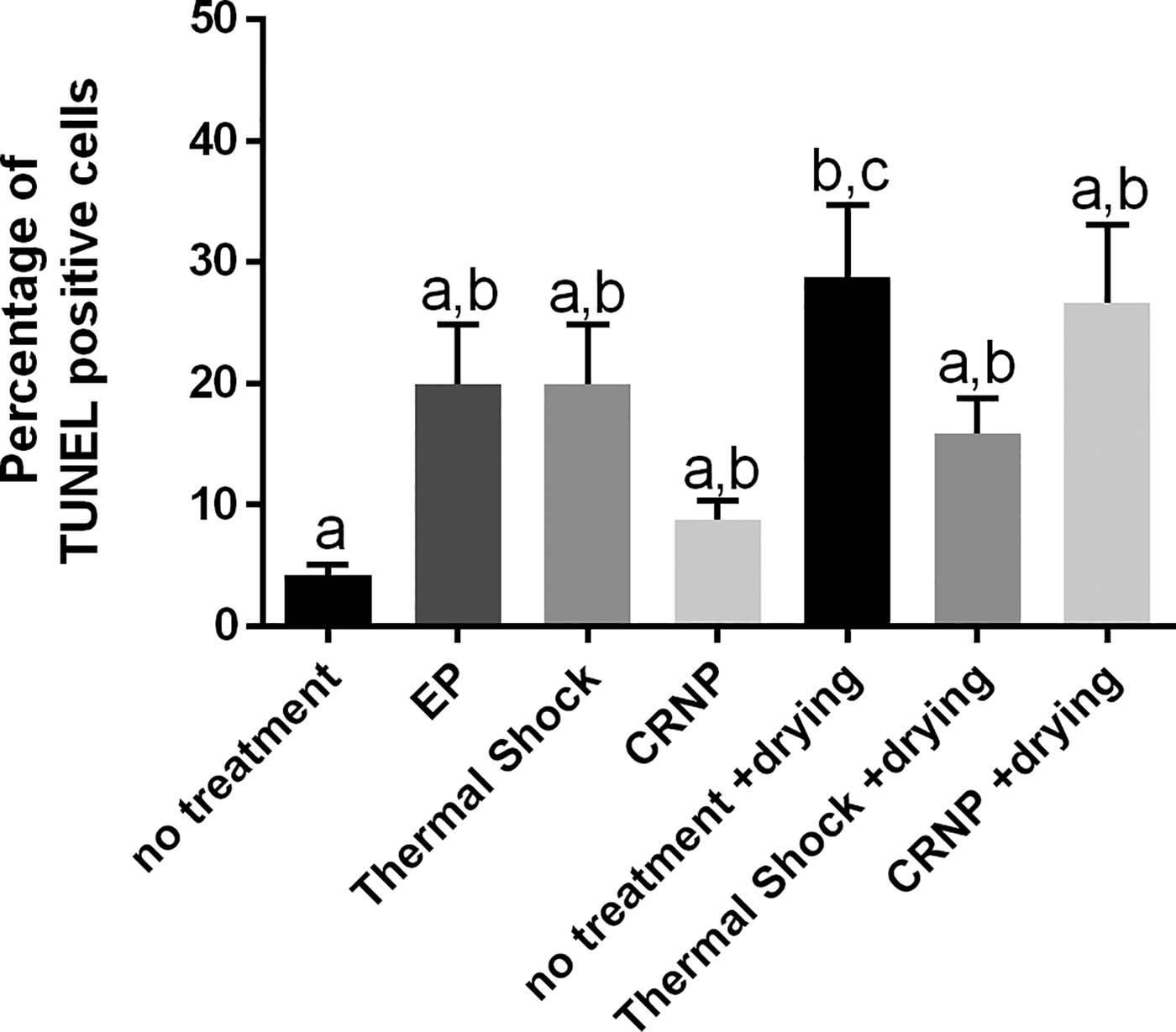
Percentage of cells (mean ± SEM) positive for DNA damage after each of the trehalose-loading treatments only or after each of the trehalose-loading methods, drying, and immediate rehydration. The electroporation followed by drying was excluded from analysis due to insufficient numbers of recoverable cells (<100 cells). Different letters above the bars indicate statistical significance (p < 0.05), and bars sharing the same letters are not statistically different from one another. EP, electroporation; CRNP, cold-responsive nanoparticles.
Effect of different trehalose-loading methods on proliferative ability after passive air-drying and rehydration
Cells were assessed for proliferating ability after reaching the dry state and immediate rehydration (no storage) for each of the trehalose-loading methods. The number of recovered cells was very low (<15 cells per method) and likely due to the additional cell handling steps that the assay required (i.e., fixing, permeabilizing, washing). However, results of this assay indicated that only a small subpopulation of cells was able to reach the dry state and maintain their proliferative ability.
Effect of different trehalose-loading methods on cell viability after passive air-drying and different dry storage conditions
On Day 0 of storage, immediately after reaching the dry state, all conditions had some viable cells (Fig. 7). There were no differences between trehalose-loading methods or untreated cells (p > 0.05) due to large SEM. Cells in the EP method had the lowest viability with an average of only 4.2 ± 1.4% viable cells. Cells loaded by thermal shock had an average of 38.1 ± 28.3% of viability. CRNP loaded cells had an average of 29.2 ± 7.5% of viability. Untreated cells tended to have the highest viability with an average of 49.7 ± 17.0%.
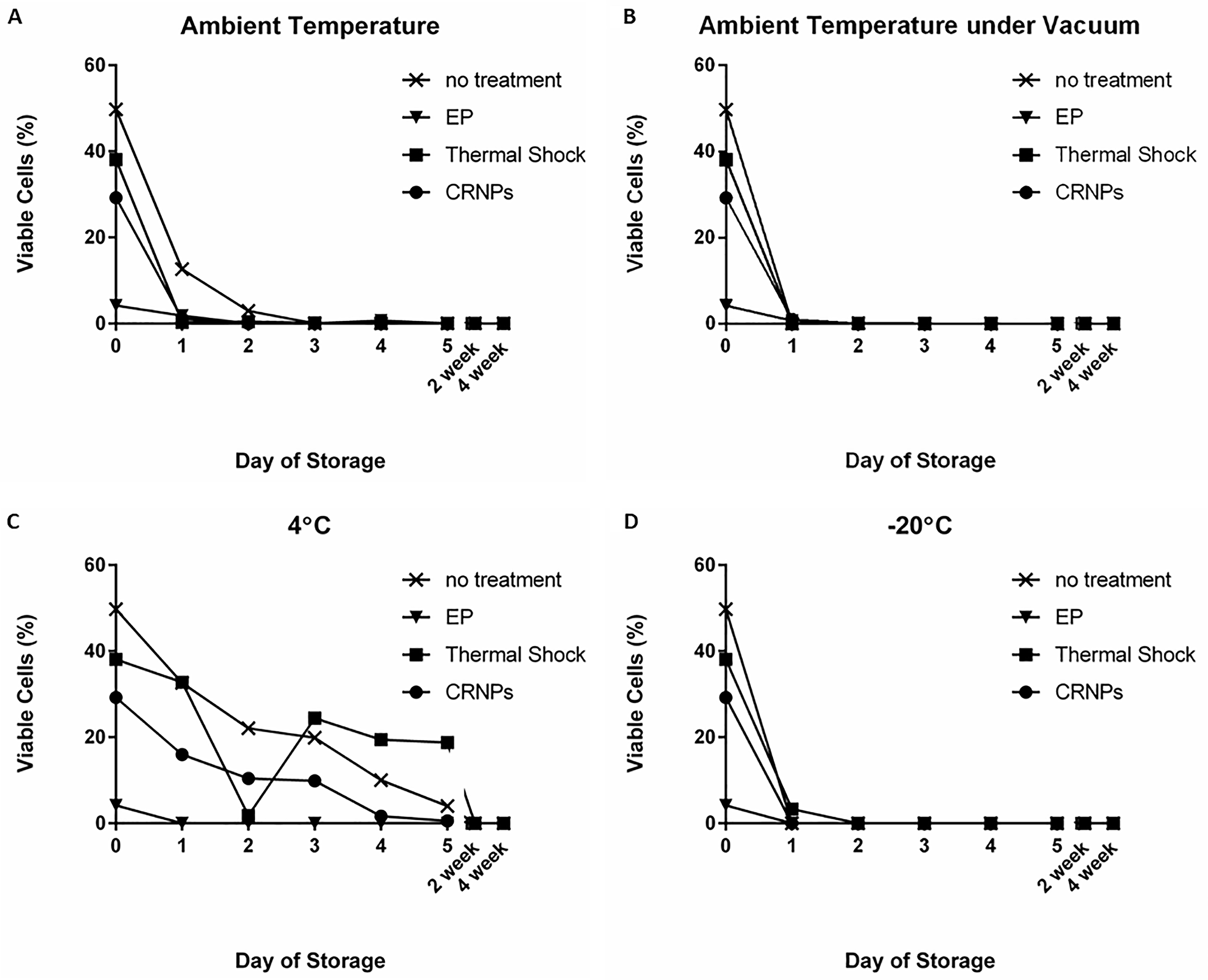
Average cell viability (shown as a percentage of the average total number of starting cells after passive drying) when stored for the initial 5 days, 2 weeks, and 4 weeks of storage at
For storage conditions at ambient temperature, cell viability decreased drastically during the first day of storage down to 12.6 ± 4.5% in the untreated cells, but this was statistically better than cells that were treated with any of the trehalose-loading methods (p < 0.01, Fig. 7A). There were no differences between any of the treatments for any other conditions or lengths of storage (p > 0.05). Cells loaded with trehalose using EP, thermal shock, and CRNPs were not viable by Day 2 of storage (Fig. 7A). Untreated cells were not viable by Day 3 of storage (Fig. 7A). During storage at ambient temperature under vacuum (Fig. 7B), the decrease in viability was even more drastic with <1% viable cells of the untreated cells remaining after Day 1, and no viable cells by Day 2 for all methods.
During storage at 4°C (Fig. 7C), cells loaded with trehalose via EP were no longer viable after 1 day of storage, but cells loaded by thermal shock, CRNP, and untreated cells had some viable cells after 5 days of storage, with the thermal shock method still having 18.8 ± 18.8% viable cells, no treatment having 4.0 ± 4.0%, and CRNP treated cells having 0.6 ± 0.6% at the Day 5 time point. During storage at −20°C (Fig. 7D), cells in the no-treatment, EP, and CRNP groups had no viable cells after any length of storage measured, and cells from the thermal shock had no viable cells after 2 days of storage through the rest of the 5-day storage period. Viability was assessed after 2 and 4 weeks of storage under each trehalose-loading method, but there were no viable cells from any treatment or storage condition at these extended time points.
Discussion
The primary objective of this project was to determine whether primary fibroblasts could be dried and rehydrated while maintaining their viability, DNA integrity, and proliferative ability. To protect the cells during the drying process, cells were first loaded with trehalose using multiple methods. Optimal EP methods had to be determined for the first time because there are no published reports about the use of EP with domestic cat primary fibroblasts. The other trehalose-loading methods (thermal shock 12 and trehalose-enclosed CRNPs4,7) could be directly adjusted from previously published reports in other species and other cell types.
In addition to being a xeroprotectant that protects cell membranes and structures during the drying process, trehalose is also a known cryoprotectant when low intracellular concentrations are achieved. 19 While both desiccation and freezing involve managing water stress, the prevention of ice crystal formation is the primary goal during cryopreservation, while achievement of a stable glassy state (prevention of crystallization) is the goal of desiccation protection. 19 Although it depends on cell type, up to 10 times less intracellular trehalose is needed to preserve cells during freeze-drying (20–25 mM) than is needed for cryopreservation (150–200 mM). 16 To ensure that each of the trehalose-loading methods was getting a minimal protective amount of trehalose into the cells, and since cryoprotective levels of intracellular trehalose were higher than those needed for protection during drying, viability after freezing and thawing was informative. Trehalose loaded by EP and thermal shock conveyed protective effects, while the CRNP did not. We hypothesized that the CRNPs were likely delivering the lowest amount of trehalose into the cells because of the slower process. There was still a variation in the percentage of cells that survived when loaded with CRNPs, which tended to be better than the untreated controls. Increased sample sizes might have achieved statistical significance, but the current intracellular levels of trehalose were not likely biologically significant. Regardless, future studies are needed to determine whether there is a more optimal way to use CRNPs to load primary fibroblasts, such as using a higher concentration of trehalose during the CRNP synthesis, using a higher concentration of CRNPs during the incubation period, and/or extending the incubation period. Importantly, less trehalose is needed to protect cells during drying than is needed during freezing, since the concentration of intracellular trehalose will increase as water is removed from the cell. 20 We therefore interpreted these results as indicating that there might be a protective effect with the amount of trehalose being loaded using the EP and thermal shock treatments, and potentially with CRNPs as well.
The drying method for this study was passive air-drying, which allows the cells to dry slowly over a period of several days.11,12,15 This is different than many other techniques that rapidly dry the cells in minutes or hours, such as microwave-assisted drying and freeze drying.7,10,21,22 In a pilot study, we exposed cells to microwaves; however, viability was lost before the cells reached the dry state. This was likely due to the increasing concentrations of solutes in the drying solution that caused severe osmotic stress to the cells, similar to the osmotic stress that is caused to cells by freezing injury.23,24 This method of slow, passive air-drying after removal of the culture medium may prevent acute osmotic stress prior to the stress of drying. Interestingly, this method even allowed for cells that were untreated (not loaded with trehalose) to survive drying and immediate rehydration. Surprisingly, untreated cells outperformed cells loaded via EP, thermal shock, and CRNPs when immediately rehydrated after reaching the dry state, and also after several days of storage at ambient temperature. One explanation for this unexpected finding is that the trehalose-loading methods used in this study, EP, thermal shock, and CRNPs, may cause varying degrees of cell damage and stress. Optimized EP led to 50%–60% viability before the drying process, which means that 40%–50% of nonviable cells likely exhibited membrane damage, lipid peroxidation, and damage to membrane-embedded proteins. 25 Both the thermal shock and CRNP methods subjected the cells to a cold treatment for a period of time, which can also induce a shock response in cells. 26 This cellular response to cold treatment may put them in a more vulnerable state than untreated cells when they go into the drying process. Another explanation could be that the slow process of drying, along with the variation in ambient temperature and relative humidity, is causing the trehalose to crystallize instead of allowing it to reach a stable glassy state. If that is the case, the crystal structures could be damaging cell membranes and structures, like the damage caused by ice crystals. Future studies will aim to perform this slow drying method in an environment where the temperature and relative humidity can be controlled. Additional experiments under more consistent conditions may also improve the statistical power of our results by reducing variance in the outcomes after drying. Additionally, future studies utilizing slow drying in a controlled environment should use analytical techniques to confirm whether the trehalose is drying into an amorphous glassy matrix that offers protection or a crystalline state, which can be damaging to the cells.
There was no difference in the proportion of cells with positive TUNEL staining between any of the treatments alone and untreated cells, indicating that the trehalose-loading methods alone were not affecting double-strand breaks of DNA. The number of cells recovered after EP and drying was too small to accurately assess with this assay so that method was excluded, but there were no differences between any of the other treatments, including untreated cells, after drying. The only statistical difference detected was between untreated cells before drying (fresh control) and cells that were not treated and then dried. These results suggest that the untreated cells have the most DNA damage after drying, which is in line with our hypothesis that the trehalose acts as a protective agent to stabilize cell structures, including DNA, during the drying process. These results are important if this drying method is used to preserve and store genetically valuable specimens for biomedical or wildlife conservation purposes, where the DNA could be the most valuable part of the stored specimen and used for downstream genetic studies or assisted reproductive technologies such as cloning or in vitro gametogenesis.2,27 We acknowledge that there are already several reported methods of preserving DNA integrity at ambient temperatures, including ethanol, lysis buffer, and dimethyl sulfoxide (DMSO) salt solution.28–30 However, these methods do not allow to recover live and functional cells, which limits the potential downstream uses to techniques that rely on DNA sequencing information only and not stem cell technologies, for instance.
Although viable cells were recovered 30 minutes after drying and rehydration, we acknowledge that only a small percentage of the starting number of cells was recovered, and that cell death may be even more prevalent at a later time point due to damage caused to the cells that cannot be recovered. We therefore wanted to make sure that, even though viable cells remained based on our trypan blue exclusion assay, they also maintained a proliferative ability. Live cells recovered after rehydration (from the same pool of cells that were used for the trypan blue exclusion assay for viability) were able to reattach to culture dishes and continue to form small colonies if they were kept in culture conditions. The number of recovered cells was smaller than the typical seeding density, which caused the growth to be slow. Unfortunately, the number of cells recovered from each group was too low to perform accurate statistical tests. However, this assay revealed that there was a small subpopulation of cells that were able to continue proliferating after being dried and rehydrated. These encouraging results indicate that it might be possible to culture and expand these surviving cells so that a much larger population of cells can be recovered. This process would actually select cells that are most adapted to surviving in the dry state, and a higher percentage of these expanded cells might survive if they were to be dried a second time. Determining the mechanisms by which this small subset of cells is able to respond so well to the stress of drying and rehydration, especially when so many others do not, is an area of interest for future studies so that the number of recoverable cells after drying can be increased.
After determining that cells remained viable after reaching the dry state and some even remained viable after 2 days at ambient temperature storage, other storage conditions were assessed to see if any could extend this viability even further. Previous reports in human mesenchymal stem cells found that vacuum storage prolonged cell physiology; 11 however, cat primary fibroblasts were not viable after only 1 day of storage under vacuum. Next, storage without vacuum at 4°C and −20°C was also assessed. These results, combined with the ambient temperature storage, suggested that while loading cells using CRNPs and EP might confer some protective effect from the trehalose, they were somehow less tolerant of the drying process than untreated cells. We hypothesized that the treatments caused either too much cellular death, as was probably the case with the EP method killing between 40% and 50% of the cells before the drying process even starts, or caused cellular stress at a level that was detrimental to the cells’ responses to the drying process. 25 Of the trehalose-loading methods evaluated in this study, thermal shock might have caused the least amount of stress to the cells because they retained the highest viability for the longest period of time compared with the other treatments, while these cells still did not respond to drying as well as the untreated cells. Again, we hypothesized that the known cellular stress responses that are induced by cold shock 26 put cells in a more vulnerable state so they did not perform as well as untreated and unstressed cells. Our findings suggest that the untreated (and unstressed) cells were most capable of surviving the drying and rehydration process; however, since these cells did not contain trehalose, they did not have any of its protective effects during the dry storage period, and therefore cells could not be maintained in the dry state for long periods of time. Storage at 4°C was able to maintain viability through the initial 5-day period, but there were no viable cells even in this condition after 2 weeks of storage. Future studies will focus on determining how to reduce stress on the cells during the trehalose-loading processes, and to determine if additional factors need to be controlled during the storage period, such as reducing the oxygen concentration (by a method other than vacuum, which may cause physical/mechanical stress on cell structures) to reduce oxidative stress on the cells during the storage period. The ultimate goal of this study was to preserve viable cells in a dry state for the long term, but we were unable to recover viable cells after only 2 weeks of storage. Future studies will be conducted to optimize storage conditions that maintain viability for much longer periods, which would be compatible with biobanking practices where cells are stored for much longer periods of time. DNA integrity and proliferative ability will also have to be assessed once storage conditions are improved.
The findings of this study serve as a foundation for future studies to continue exploring dry preservation as a sustainable alternative for preserving fibroblasts as a genetic resource. We have shown that cells are able to maintain their viability, DNA integrity, and proliferative ability after reaching a dry state and being rehydrated, but only remain viable in the very short term. There may be practical benefits to this already, including shipping/transportation methods that are less cumbersome than maintaining cold storage, which has already been reported in other cell types.11,12 Additionally, this could be used to collect samples for genetic testing or sequencing that can be performed in the short term. This would be mostly beneficial for transporting specimens without temperature constraints or the need for specialized equipment. While these are still beneficial uses of keeping biological samples at ambient temperatures for short periods of time, we realize the value of longer-term storage without needing ultracold temperatures would be much greater, but it requires more work before that goal can be reached. We will continue exploring the underlying mechanisms as we work to find storage conditions that can prolong viability beyond the short term. Ultimately, storage at ambient temperature will reduce costs and increase sample storage capacity across multiple disciplines including human and veterinary medicine and also has implications for wildlife conservation efforts.
Authors’ Contributions
D.M.S.: Conceptualization, formal analysis, investigation, methodology, writing—original draft, writing—review and editing, visualization, data curation, and funding acquisition. A.J.: Formal analysis, investigation, and writing—review and editing. O.S.: Formal analysis, investigation, and writing—review and editing. P.C.: Conceptualization, formal analysis, resources, funding acquisition, supervision, writing—review and editing, methodology, and project administration.
Ethical Considerations
Not applicable. Experiments described did not require the approval of the Animal Care and Use Committee of Smithsonian’s National Zoo and Conservation Biology Institute because reproductive tracts were collected at local veterinary clinics as byproducts of owner-requested routine ovariohysterectomies.
Data Availability
Full data spreadsheet can be made available upon request.
Supplemental Material
sj-docx-1-bpb-10.1177_19475535261463415 — Supplemental material for Response of Domestic Cat Primary Fibroblasts to Trehalose-Loading Methods, Air-Drying, and Storage Conditions at Non-freezing Temperatures
Supplemental material, sj-docx-1-bpb-10.1177_19475535261463415 for Response of Domestic Cat Primary Fibroblasts to Trehalose-Loading Methods, Air-Drying, and Storage Conditions at Non-freezing Temperatures by Danielle M. Sosnicki, Abigail Jensen, Olivia Spicer, and Pierre Comizzoli
Footnotes
Acknowledgments
The authors thank Dr. Keiko Antoku and her staff for the collection of domestic cat reproductive tracts, and Dr. James Shamul of the He Lab at the University of Maryland for his assistance with characterizations of CRNPs.
Author Disclosure Statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding Information
This work was funded by a Smithsonian Fellowship, the JoGayle Howard Revocable Trust, and a Smithsonian Scholarly Studies grant.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.