
Abstract
Background:
To investigate the effects of storage time and temperature on the quality of fecal samples and to provide a reference for clinical laboratories and biobanks in formulating sample storage operation guidelines.
Methods:
Fresh fecal samples were collected from healthy volunteers and immediately aliquoted into aliquots. Different temperature and time gradients were established to simulate common pre-analytical storage processes in clinical practice, with samples snap-frozen in liquid nitrogen immediately after collection as the control group. 16S rRNA gene sequencing and untargeted lipid metabolomics were employed to determine changes in microbial diversity, species abundance, and metabolite concentrations under different storage conditions, and sample quality was evaluated based on these indicators.
Results:
Storage at 4°C significantly minimized fluctuations in α-diversity indices, with the most pronounced protective effect observed within 2–4 hours; beyond 4 hours, changes in microbial community structure intensified. β-diversity analysis revealed that 4°C storage delayed the increase in microbial dissimilarity between samples and the liquid nitrogen-frozen control group, among which samples stored for 2–4 hours exhibited the highest similarity to the control. At the phylum level, the abundance of Firmicutes increased significantly after 6 hours of storage at room temperature, while 4°C storage effectively delayed this change. Metabolomic analysis identified more metabolomic differences (including bile acids and amino acids) in samples stored at room temperature, whereas only minor changes in fatty acid metabolites were observed at 4°C. 3β-hydroxy-5-cholenic acid exhibited a continuous upward trend with prolonged storage at both temperatures, suggesting its potential as a biomarker for evaluating sample storage quality.
Conclusion:
Short-term storage at 4°C (≤4 hours) can effectively delay the quality degradation of fecal microbial communities and metabolites, making it the optimal transitional storage strategy when immediate liquid nitrogen freezing is not feasible in clinical practice. These findings provide critical experimental data for the establishment of standardized fecal sample storage protocols.
Introduction
The gut, as the largest microecosystem in the human body, harbors a complex community of intestinal microbiota that function as an “invisible organ” involved in key physiological processes, such as metabolism, immune regulation, and nutrient absorption. Its functional significance extends throughout the entire life cycle.1,2 Through co-evolution with the host, these microbiota establish a dynamic balance within the intestinal microenvironment, and the stability and diversity of their community structure are critical for sustaining human health. 3
The association between gut microbiota and human health extends beyond traditional digestive functions, emerging as a key clue for deciphering the pathogenesis of various diseases.4,5 Studies have confirmed that microbial dysbiosis is not only directly linked to gastrointestinal disorders such as irritable bowel syndrome and inflammatory bowel disease but also modulates the risk of metabolic syndrome, diabetes, neurodegenerative diseases, and even tumors via multiorgan cross-talk pathways, including the gut–brain axis and gut–liver axis.6–8 A healthy gut microbiota acts as a crucial barrier for maintaining metabolic balance and immune homeostasis, while the disruption of microbial communities may act as a potential trigger for various diseases, offering new avenues for early disease screening and targeted therapeutic intervention.
Given the intimate link between gut microbiota and human health, fecal samples, as direct carriers of intestinal microbiota, have gained growing clinical significance.9,10 Compared with invasive detection modalities, fecal samples offer distinct advantages such as noninvasiveness, ease of collection, and the ability to provide a comprehensive reflection of gut microecological status. They are widely used in the screening and diagnosis of digestive diseases, etiological identification of intestinal infections, and risk stratification of metabolic diseases and have shown substantial potential in early tumor screening, emerging as an indispensable noninvasive diagnostic tool in clinical practice.11,12
Notably, the quality of fecal sample preservation directly determines the accuracy and reliability of test results, serving as a core prerequisite for their clinical utility.13,14 Biomarkers in fecal samples, such as bacteria and nucleic acids, are susceptible to environmental variables, including temperature, humidity, and storage duration, which may induce the degradation of these biomarkers or alter the composition of the gut microbial community—ultimately resulting in the distortion of test outcomes. Therefore, optimizing sample storage protocols and selecting appropriate preservation media and conditions are critical for ensuring the validity of gut microbiota-related assays and facilitating the widespread clinical application of fecal testing technologies. 15
Previous studies have demonstrated significant differences in the composition of Bacteroidota and Firmicutes in fecal samples following 15 minutes of storage at room temperature, suggesting that such samples should be frozen within 15 minutes after defecation. 13 However, in practical clinical settings, achieving freezing or testing within this 15-minute window post-sample collection is highly challenging and thus difficult to implement on a large scale. Therefore, establishing a feasible and operable protocol for fecal sample collection and preservation holds considerable clinical significance.
This study integrated 16S rRNA gene sequencing and untargeted metabolomics approaches to systematically evaluate the effects of different storage conditions on the stability of fecal microbial diversity and metabolic profiles. It aimed to elucidate the attenuation dynamics of biological information within the clinical testing time window and provide an experimental basis for formulating standardized sample acceptance criteria. The findings are expected to address the research gap of multiomics validation in current preservation guidelines and facilitate the standardization of the entire workflow from fecal sample collection to downstream analysis.
Materials and Methods
Study subjects
Healthy volunteers were recruited as study participants, and their fecal samples were collected for investigating the effects of storage parameters on fecal microbiota under controlled storage conditions (H1–H11).
Inclusion criteria for healthy volunteers were as follows:
No history of cardiovascular or endocrine diseases, including coronary heart disease, hyperlipidemia, or diabetes mellitus. No history of digestive disorders (e.g., diarrhea and gastroenteritis) within the preceding week. No episodes of binge eating or excessive consumption of greasy, high-calorie foods within the preceding week.
This study was approved by the Ethics Committee of Beijing Anzhen Hospital (KS2023001), and all participants provided written informed consent prior to enrollment.
Sample collection and storage conditions
Fresh fecal samples were collected from 11 volunteers using sterile fecal collection containers. All participants were required to fast for 12 hours prior to sample collection, and the perianal region was thoroughly disinfected with 75% ethanol before sample procurement. Samples from volunteers were collected in 25 mL sample containers (SARSTEDT, Shanghai, China). The samples were then immediately aliquoted into 2.5 mL pre-aliquoted cryogenic vials (BaiBan, Hangzhou, China) and stored promptly under the designated preservation conditions as planned. Each sample was aliquoted into sterile cryopreservation tubes and subjected to the following storage conditions:
Control group (CON): Snap-frozen in liquid nitrogen immediately after collection. RT1: Held at room temperature for 2 hours, followed by snap-freezing in liquid nitrogen. RT2: Held at room temperature for 4 hours, followed by snap-freezing in liquid nitrogen. RT3: Held at room temperature for 6 hours, followed by snap-freezing in liquid nitrogen. FT1: Held at 4°C for 2 hours, followed by snap-freezing in liquid nitrogen. FT2: Held at 4°C for 4 hours, followed by snap-freezing in liquid nitrogen. FT3: Held at 4°C for 6 hours, followed by snap-freezing in liquid nitrogen.
Fecal DNA extraction
Genomic DNA was extracted from fecal samples using the FastPure Stool DNA Isolation Kit (MJYH, Shanghai, China), and DNA concentration and purity were determined using a NanoDrop2000 ultraviolet spectrophotometer (Thermo Scientific, MA, USA).
First, 250 mg of glass beads were accurately weighed and transferred into a 2.0 mL centrifuge tube. Subsequently, approximately 200 mg of fecal sample was added to the same tube, followed by the sequential addition of 750 μL of Lysis Buffer A and 90 μL of Lysis Buffer B. The mixture was homogenized twice using a mill at 45 Hz for 300 seconds each time to ensure sufficient lysis of the sample.
After homogenization, the centrifuge tube was centrifuged at 14,000 rpm for 5 minutes. A total of 500 μL of the supernatant was carefully transferred to a new 1.5 mL microfuge tube, and 100 μL of Purification Buffer I as well as 100 μL of Purification Buffer II were added to the supernatant. It should be noted that the bottle of Purification Buffer II must be vigorously shaken or vortexed to form a uniform suspension prior to use. The tube was inverted 30–50 times to mix the contents thoroughly, and vortexing was strictly avoided during this process.
The mixture was then centrifuged at 14,000 rpm for 5 minutes, after which 400 μL of the supernatant was transferred to a new 1.5 mL microfuge tube. Next, 400 μL Binding Buffer and 60 μL Magnetic Beads were added to the supernatant; prior to adding the Magnetic Beads, they should be thoroughly resuspended. The tube was vortexed to mix well and then incubated at room temperature for 3 minutes to facilitate the binding of DNA to the magnetic beads.
Following incubation, the tube was placed on a magnetic separator for 3 minutes to magnetize the Magnetic Beads. The clarified supernatant was carefully discarded, taking care to avoid aspirating the settled binding beads. Then, 500 μL of Washing Buffer I was added to the tube, and the Magnetic Beads were resuspended by vortexing. The tube was again placed on the magnetic separator for 3 minutes, and all clarified supernatant was discarded cautiously to prevent the loss of Magnetic Beads.
Subsequently, 800 μL of Washing Buffer II (to which a specified volume of anhydrous ethanol must be added before use) was added to the tube, and the Magnetic Beads were resuspended by vortexing. The tube was placed on the magnetic separator for 1 minute, and the clarified supernatant was discarded carefully. The same operation was repeated with 800 μL of Washing Buffer III (also requiring the addition of a specified volume of anhydrous ethanol before use): After resuspending the Magnetic Beads by vortexing, the tube was placed on the magnetic separator for 1 minute, and the supernatant was discarded cautiously. This washing step with Washing Buffer III was repeated once more to ensure thorough removal of impurities.
After the final washing, any residual liquid in the tube was removed using a pipette, and the Magnetic Beads were air-dried at room temperature for 3–5 minutes. It is critical to ensure complete removal of residual ethanol, as it may interfere with subsequent downstream experiments. Then, 100 μL of Elution Buffer was added to the tube, and the dried Magnetic Beads were resuspended by vortexing, followed by incubation at 56°C for 5 minutes to elute the DNA from the Magnetic Beads.
Finally, the tube was placed on the magnetic separator for 3–5 minutes to re-magnetize the Magnetic Beads. The purified DNA supernatant was carefully transferred to a new 1.5 mL tube, with strict avoidance of aspirating the settled binding beads, completing the entire fecal DNA extraction process.
16S rRNA analysis
The V3-V4 variable region of the 16S rRNA gene was amplified by PCR using barcode-tagged forward primer 338 F (5′-ACTCCTACGGGAGGCAGCAG-3′) and reverse primer 806 R (5′-GGACTACHVGGGTWTCTAAT-3′). Alpha diversity indices such as Chao and Shannon were calculated using mothur software, and intergroup differences in alpha diversity were analyzed by the Wilcoxon rank-sum test. Principal Component Analysis (PCA) based on distance algorithms was used to assess the similarity of microbial community structures between samples, and a PERMANOVA nonparametric test was performed to analyze the significance of intergroup differences in microbial community structures.
Untargeted metabolomics analysis
Samples were analyzed by LC-MS/MS using the Thermo Scientific UHPLC-Q Exactive HF-X system. Raw LC-MS data were imported into the metabolomics software Progenesis QI (Waters Corporation, Milford, USA) for baseline filtering, peak detection, integration, retention time correction, and peak alignment, resulting in a data matrix containing retention time, mass-to-charge ratio, and peak intensity. MS and MS/MS spectra were matched against public metabolomic databases (HMDB: http://www.hmdb.ca/;Metlin: https://metlin.scripps.edu/) and a self-built database by Majorbio to obtain metabolite information.
Statistical analysis
All gene sequencing data were analyzed on the Majorbio Cloud Platform (https://cloud.majorbio.com). Statistical analysis was performed using SPSS 22.0 software. Normally distributed quantitative data were expressed as mean ± standard deviation (χ ± s), while nonnormally distributed data were expressed as median (interquartile range). An independent samples t-test was used for the comparison of intestinal microbial alpha diversity, PERMANOVA (Permutational Multivariate Analysis of Variance) test for beta diversity, and a Wilcoxon rank-sum test for microbial abundance. The significance level α was set at 0.05. All statistical analyses were corrected for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) procedure.
Results
Interindividual differences are greater than intragroup differences induced by storage conditions
PCA based on 16S rRNA sequencing data showed distinct individual-specific clustering patterns of fecal microbial communities under different storage conditions (Fig. 1A). Each point represents a single fecal sample, with samples from the same volunteer (H1–H11) marked with the same symbol.
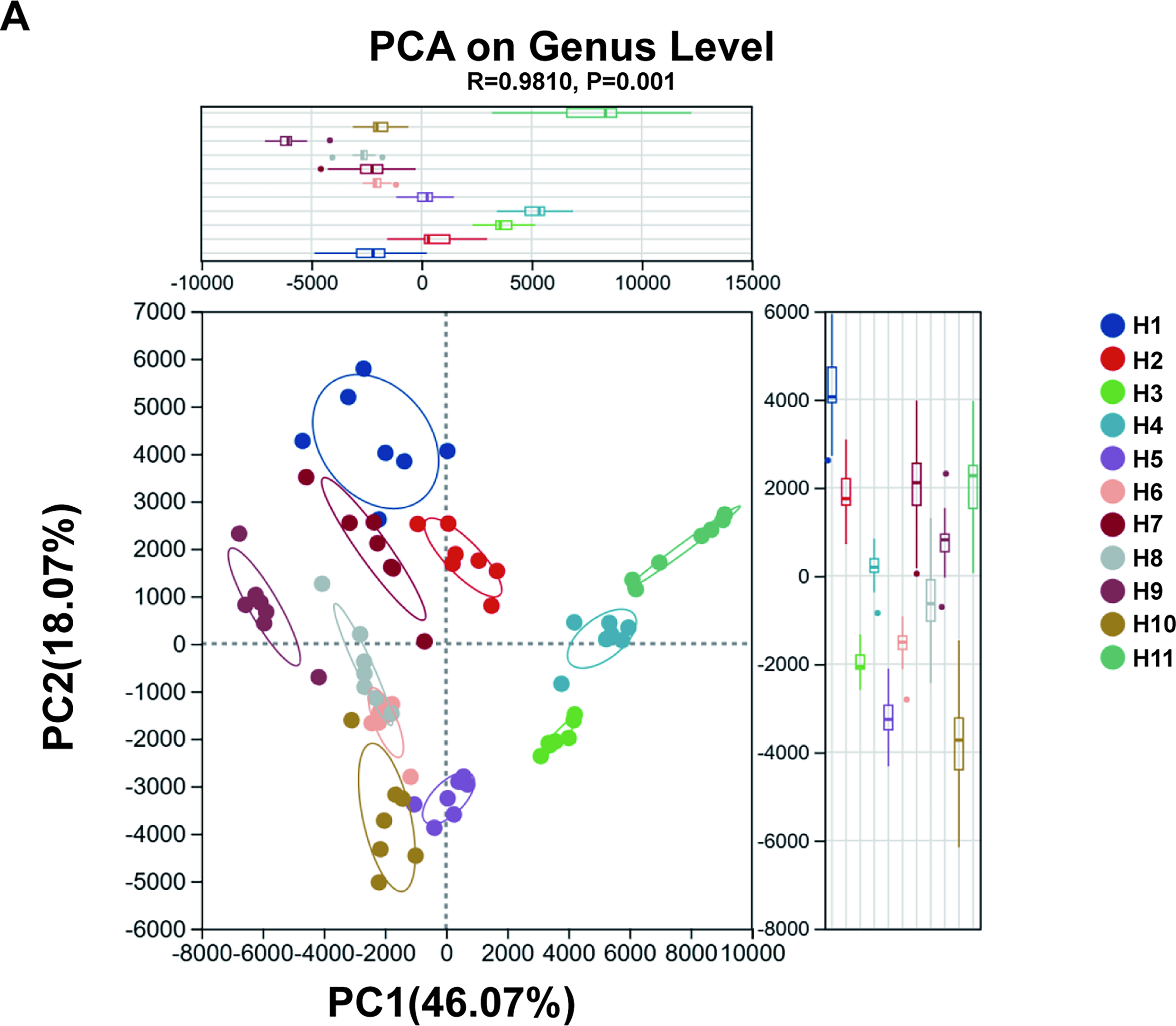
Principal Component Analysis (PCA) of fecal microbial communities at the genus level based on 16S rRNA gene sequencing data
Despite seven different storage treatments, seven samples from the same volunteer preferentially clustered into independent groups in the PCA space, indicating that inherent interindividual differences in gut microbiota were far greater than variations introduced by storage conditions. This phenomenon suggests that host individual characteristics (e.g., genetic background and dietary habits) play a dominant role in shaping gut microbial composition, with an impact strong enough to override microbial community fluctuations caused by different preservation methods.
Notably, seven samples from the same volunteer showed a certain degree of dispersion within clusters, which may reflect the differential effects of storage conditions on the abundance of specific microbial taxa. For example, oxygen-sensitive or nutrient-dependent genera may undergo selective changes under treatments such as freezing temperature, freeze-thaw cycles, or preservative exposure. However, the magnitude of such variations induced by storage conditions was relatively limited and did not disrupt the overall clustering characteristics of individual samples, indicating that the core microbial community maintained high stability during experimental treatments.
Alpha diversity evaluates sample species richness
This study analyzed the variation characteristics of common alpha indices under different storage conditions (Fig. 2). Due to interindividual differences, no significant differences were observed in the absolute values of alpha diversity among groups. Therefore, the percentage change of alpha indices relative to the CON group was calculated with the CON group as the baseline.
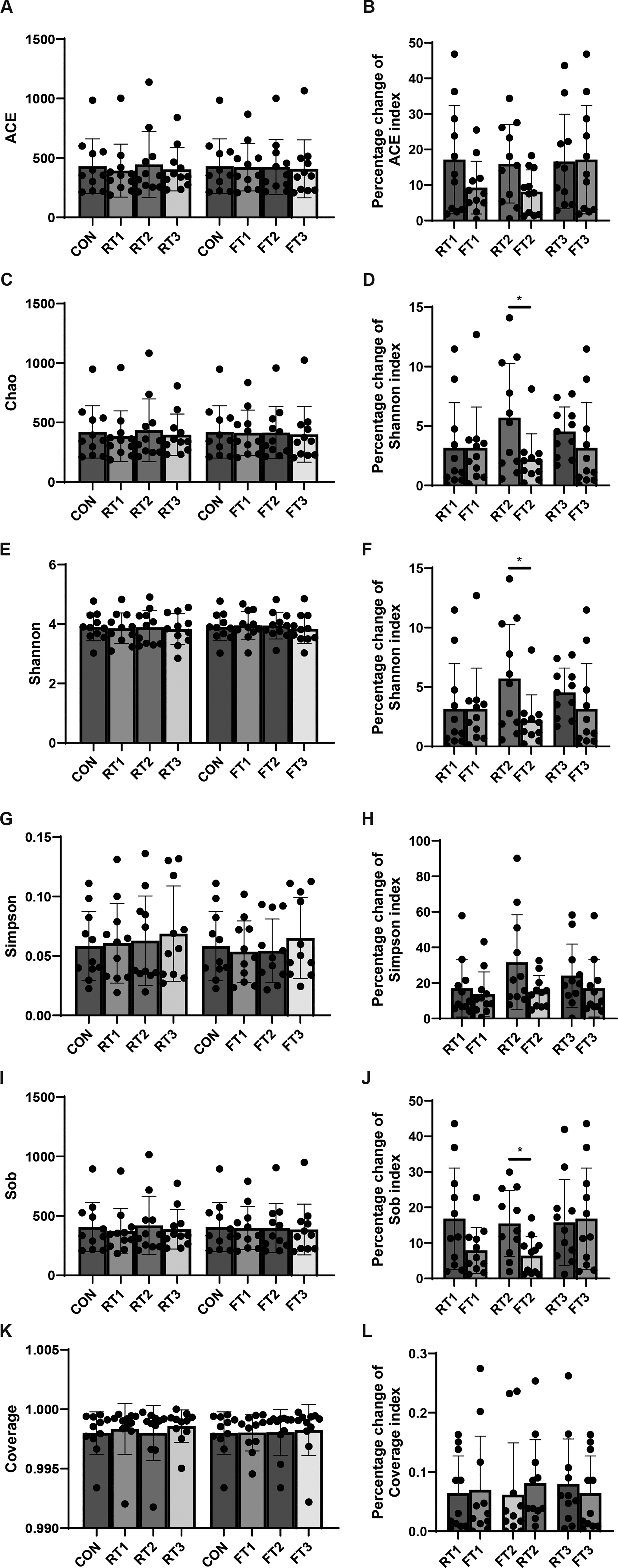
Alpha diversity analysis of fecal samples under different storage conditions
The Chao index is primarily used to estimate the true total number of species in a sample, with particular emphasis on capturing unobserved rare species (Fig. 2C,D). At room temperature, the Chao index of fecal samples from 11 volunteers showed significant changes after 2 hours of storage. Compared with room temperature storage for 2 and 4 hours (RT1 and RT2 groups), 4°C storage for 2 and 4 hours (FT1 and FT2 groups) significantly reduced the percentage change of the Chao index relative to the CON group.
The Shannon index and Sob index showed the same trend: compared with the RT1 and RT2 groups, the percentage changes of these indices relative to the CON group were significantly lower in the FT1 and FT2 groups (Fig. 2E,F, 2I, 2J). While each comparison showed significance in the unadjusted t-tests, none retained significance following Benjamini–Hochberg FDR correction (adjusted p > 0.05).
In addition, although there were no statistical differences in the ACE(Abundance-based Coverage Estimator) index and Simpson index between the FT2 group and the CON group, a relatively obvious decreasing trend was still observed, which may be related to the high heterogeneity of fecal samples (Fig. 2A,B, 2G, 2H).
These results indicate that 4°C storage can significantly reduce the magnitude of changes in sample microbial diversity, but the cryoprotective effect can only be maintained for 4 hours. Beyond 4 hours, changes in the fecal microbial community will increase significantly.
Beta diversity analysis
Hierarchical clustering analysis showed that fecal samples from the same volunteer clustered into one group, indicating high similarity of microbial communities within groups (Fig. 3). However, the length of clustering branches within the same volunteer group varied with storage conditions. By comparing samples under different storage conditions within each group with those under control conditions, the sample distances value was calculated; a higher value indicated greater microbial difference (Fig. 3A). The results showed that the distance index between the RT1 group and the CON group was 0.1309 ± 0.0424; 0.1827 ± 0.0615 for the RT2 group; 0.1810 ± 0.0503 for the RT3 group; 0.1506 ± 0.0410 for the FT1 group; 0.1489 ± 0.0534 for the FT2 group; and 0.1845 ± 0.0410 for the FT3 group (Fig. 3B).
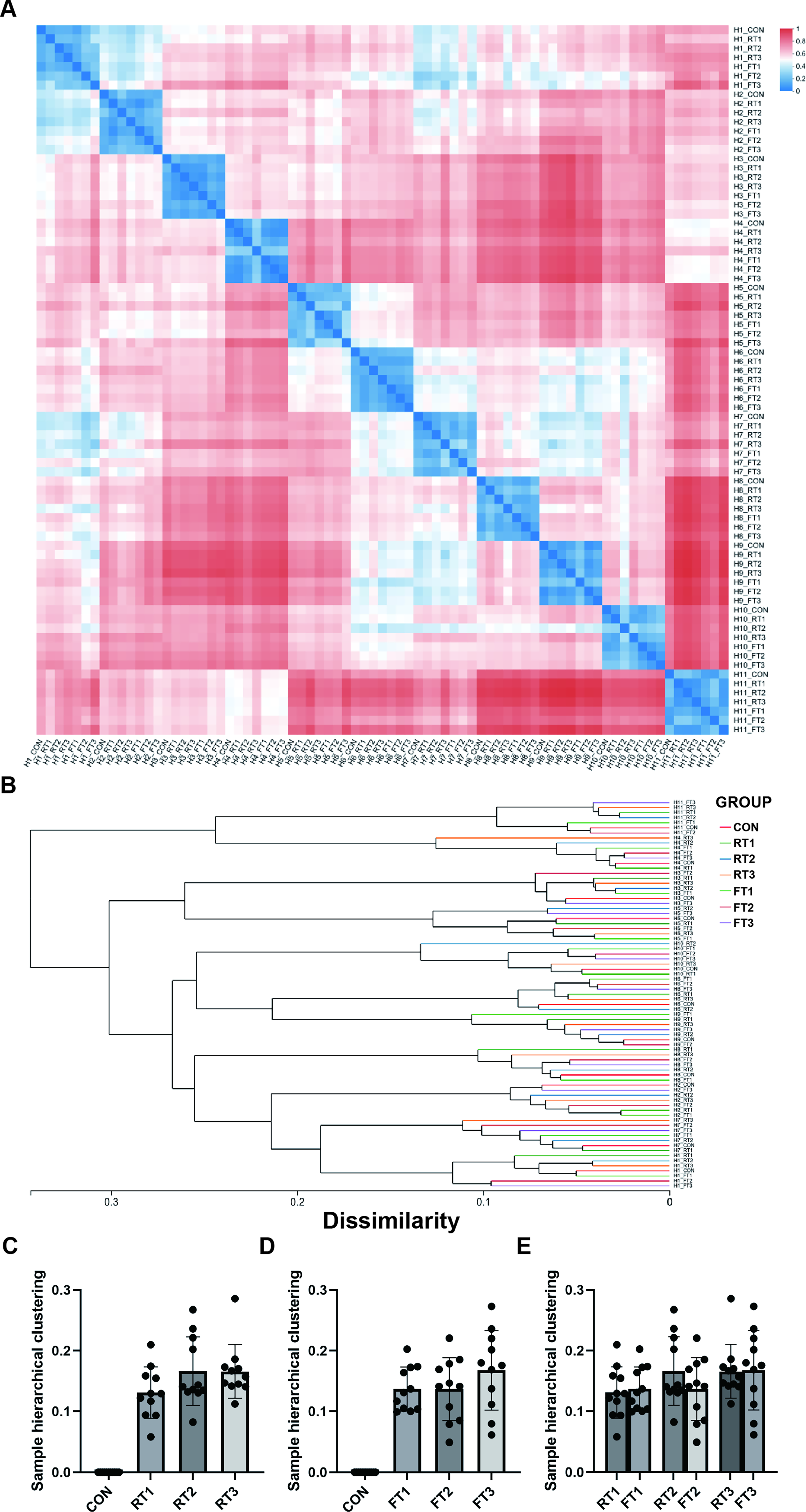
Hierarchical clustering analysis of fecal microbial communities based on 16S rRNA gene sequencing data
The results indicate that under the same temperature condition, the longer the storage time, the larger the distance value between samples and the control group, and the more significant the microbial difference. Although 4°C storage cannot completely avoid this issue, it can significantly delay the increase in sample distance. If sample processing is completed within 4 hours, the impact of microbial changes caused by room temperature storage on sample quality can be minimized.
Analysis of sample microbial abundance
Analysis of changes in microbial abundance across groups showed variations in the abundance of common Firmicutes genera such as Bacteroides, Roseburia, and Blautia (Fig. 4A). However, due to interindividual differences in microbial characteristics and complex interactions between genera, no obvious pattern was observed in the changes of individual genera (Fig. 4B–U). Including Blautia (Fig. 4B), Faecalibacterium (Fig. 4C), bifidobacterium (Fig. 4D), Eubbacterium (Fig. 4E), Collinsella (Fig. 4F), Coprostasnoligenes (Fig. 4G), Agathobacter (Fig. 4H), Dorea (Fig. 4I), Subdoligranulum (Fig. 4J), Fusicatenibacter (Fig. 4K), Bacteroides (Fig. 4L), Lachnospiraceae (Fig. 4M), Roseburia (Fig. 4N), Erysipelotrichaceae (Fig. 4O), Anoerostipes (Fig. 4P), Troques (Fig. 4Q), Coprococcus (Fig. 4R), Ruminococcus (Fig. 4S), Firmicutes (Fig. 4T), and Actinobacteria (Fig. 4U). When the classification level was elevated to the phylum level, it was found that the total number of Firmicutes bacteria increased significantly in the 6-hour room temperature storage group, while 4°C storage significantly delayed this growth trend (Fig. 4T,U).
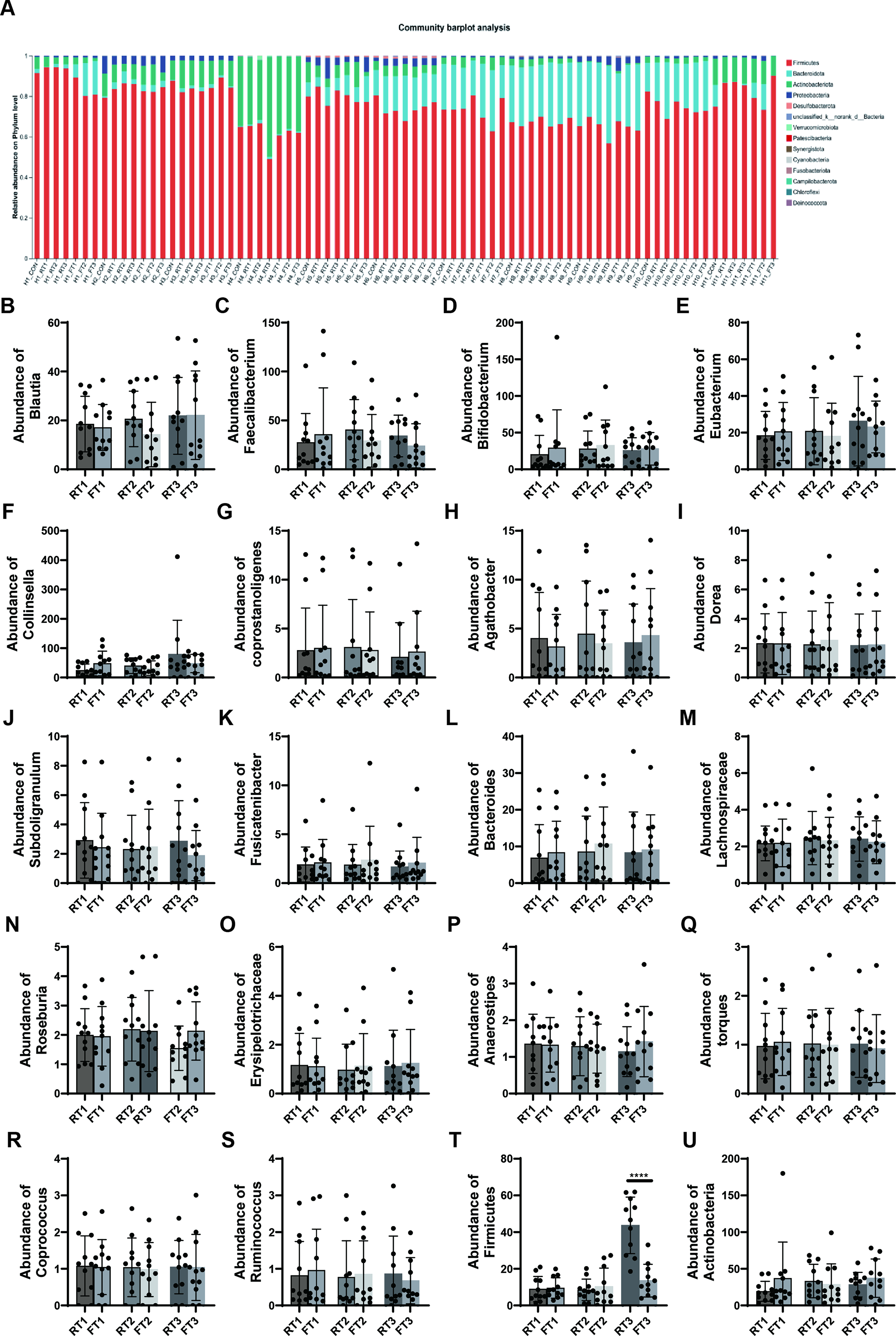
Community barplot analysis of fecal microbial composition at the phylum level, with Firmicutes as one of the dominant phyla shown
Differential metabolite screening via untargeted metabolomics analysis
In the room temperature storage group, there were numerous metabolomic differences, including 5β-cholestane-3α,7α,12α,23R,25-pentol, 3β-hydroxy-5-cholenic acid, L-proline, talatisamine, dihomo-linoleic acid, and 9,11-conjugated linoleic acid (Fig. 5A). Under 4°C storage conditions, fewer metabolites showed statistically significant changes with storage time, only 3β-hydroxy-5-cholenic acid and 9,10-dihydroxystearic acid, both of which belong to fatty acid metabolites (Fig. 5B).
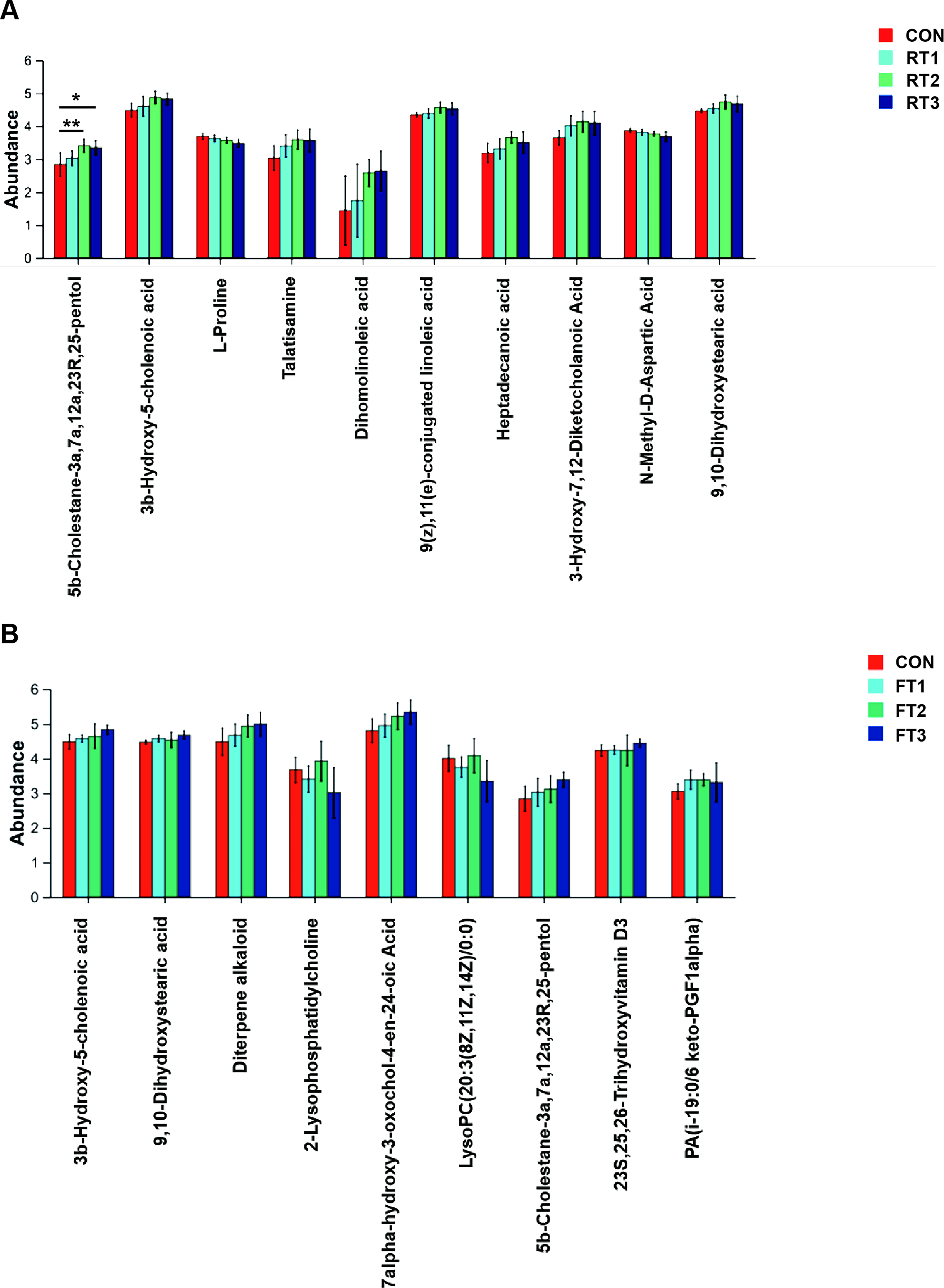
Effect of different room temperature storage durations (RT1-RT3) on the metabolite profile of fecal samples
Notably, 3β-hydroxy-5-cholenic acid showed a continuous upward trend with prolonged storage at both temperatures: Under room temperature conditions, the level was 4.485 ± 0.211 in the CON group, 4.6030 ± 0.308 in the RT1 group, 4.874 ± 0.1904 in the RT2 group, and 4.8260 ± 0.1736 in the RT3 group; under 4°C conditions, it was 4.578 ± 0.1031 in the FT1 group, 4.648 ± 0.353 in the FT2 group, and 4.837 ± 0.1241 in the FT3 group. In addition, metabolites with significant changes in different groups were mostly fatty acid metabolites, suggesting that strict standardization of sample collection procedures is required for clinical fecal samples involved in fatty acid metabolism-related detection.
Discussion
Although storage of fecal samples at −20°C or −80°C is recognized as the gold standard for sample preservation, its implementation in routine clinical practice remains challenging. 16 This study systematically investigated the effects of different storage durations at room temperature and 4°C on fecal microbiota and metabolites by employing 16S rRNA gene sequencing and untargeted metabolomics approaches. The core finding is that short-term storage at 4°C can effectively mitigate sample quality degradation, thereby providing critical data support for the standardization of operational protocols in clinical testing and biobanking facilities.
Regarding the impact of storage conditions on microbial diversity, inherent interindividual variations in gut microbiota are significantly greater than those induced by storage conditions—a finding consistent with the consensus that gut microbiota are long-term shaped by core factors such as host genetics and diet. However, storage temperature and duration still exert quantifiable effects on microbial community structure: alpha diversity indices (e.g., Chao and Shannon) exhibited notable fluctuations following 2 hours of room temperature storage, whereas 4°C storage significantly attenuated the magnitude of these fluctuations, with the most pronounced effect observed within 2–4 hours. Nevertheless, the protective effect of low temperature can only be sustained for 4 hours, beyond which microbial alterations intensify. Additionally, previous studies have demonstrated that although changes in the taxonomic composition of sample microbiota are not particularly prominent, DNA content and integrity still undergo alterations due to microbial metabolism. 17 This indicates that even in low-temperature environments, the metabolic activities and intermicrobial interactions of intestinal microorganisms can gradually reshape the community structure.
Beta diversity analysis further confirmed that 4°C storage can delay the increase in microbial dissimilarity between samples and the control group, with samples stored at 4°C for 2–4 hours exhibiting the highest similarity to the liquid nitrogen-frozen control. This finding provides a clear temporal reference for clinical testing practices: if immediate liquid nitrogen snap-freezing is not feasible, 4°C refrigeration can be employed as a transitional preservation strategy, but subsequent sample processing must be completed within 4 hours.
Metabolomic analysis revealed that the impact of storage conditions on fecal metabolites is highly class-specific, with fatty acid metabolites being the most susceptible to storage-related alterations. Among these, 3β-hydroxy-5-cholenic acid exhibited a continuous increase with prolonged storage at both room temperature and 4°C, indicating its potential as a biomarker for assessing fecal sample storage quality. The number of differential metabolites under room temperature storage was significantly higher than that under 4°C storage, including key disease-associated metabolites such as bile acids and amino acids. This finding carries important clinical implications: for metabolomic testing of fecal samples—particularly the analysis of indicators such as fatty acids and bile acids—storage conditions must be strictly regulated. Storage at room temperature for more than 2 hours may induce distortion of metabolic profiles, thereby interfering with the identification of biomarkers for diseases such as inflammatory bowel disease and colorectal cancer. This observation is consistent with the concern raised in the introduction that “attenuation of biological information masks pathological characteristics.”
The innovation of this study lies in integrating dual-omics data of microbiota and metabolites to verify the regulatory effects of storage conditions on the multiomics profiles of samples, filling the gap in single-dimensional analysis in existing studies. The results clarify the optimal operational process for clinical testing: Liquid nitrogen freezing is preferred immediately after sample collection; if immediate processing is not possible, short-term storage at 4°C (≤4 hours) is recommended, with room temperature placement or prolonged low-temperature storage strictly avoided.
For biobanks, this study provides an empirical and quantitative basis for the formulation of sample accession criteria: storage at 4°C for no more than 4 hours should be adopted as a core quality control metric for fecal sample accession. Meanwhile, detailed storage conditions should be recorded in sample information to provide reference for subsequent data interpretation. In addition, the dominant confounding role of interindividual differences in microbial structure suggests that individual baseline characteristics must be fully considered in sample storage-related studies so as to avoid masking the specific effects of storage conditions induced by interindividual differences.
This study has certain limitations: The sample size is relatively modest (11 healthy volunteers), and no diseased cohorts were enrolled; thus, the generalizability of the findings warrants further validation with an expanded sample size. Additionally, the impacts of long-term cryopreservation conditions (e.g., −20°C and −80°C) were not explored, nor were comparisons of other preservation strategies such as the addition of preservation solutions. Future research should be extended in three directions: First, expanding the recruitment of samples from diseased cohorts to investigate the impacts of storage conditions on gut microbiota and metabolites under pathological states; second, extending the storage duration to over 24 hours to analyze the long-term degradation kinetics of sample quality at different temperatures; and third, integrating the use of preservation solutions to explore optimal combinatorial sample preservation protocols. Furthermore, the correlation between microbiota and metabolite alterations can be further elucidated to establish a more comprehensive multidimensional indicator system for sample quality assessment.
In summary, this study confirms that short-term storage at 4°C is the optimal transitional preservation strategy for fecal samples. It provides a scientific basis for standardized operations in clinical testing and biobanks, facilitating the improvement of the reliability and reproducibility of gut microecology research data.
Authors’ Contributions
S.J. contributed to conceptualization, methodology, validation, formal analysis, investigation, data curation, and writing the original draft. H.T., N.J., and X.D. participated in methodology, validation, and investigation and provided critical revision of the article. X.Z. was responsible for conceptualization, resources, supervision, project administration, funding acquisition, and review and editing of the article. All coauthors have reviewed and approved of the article prior to submission.
Footnotes
Author Disclosure Statement
No potential conflicts of interest are declared.