
Abstract
Objective
Oxidative stress and endoplasmic reticulum (ER) stress play pivotal roles in disrupting the homeostasis of chondrocytes by producing catalytic proteases and enhancing chondrocyte senescence, consequently contributing to the progression of osteoarthritis (OA). Despite their close interaction, the underlying molecular mechanisms remain poorly understood. Here, we show that ER stress and oxidative stress reciprocally modulate each other to promote cartilage degradation.
Methods
Primary chondrocytes were obtained from the articular cartilage of 5-day-old C57BL/6J mice by excising distal femur and proximal tibia. Tunicamycin was applied to induce ER stress in primary chondrocytes. Surgical OA was induced in 12-week-old male C57BL/6J mice by destabilizing the medial meniscus (DMM).
Results
Tunicamycin-induced ER stress led to an increase in the production of reactive oxygen species (ROS) and catalytic proteases, including MMP13 and Adamts5, in primary chondrocytes, and it was primarily dependent on the NADPH oxidase (NOX) system. ER stress directly increased the expression of NOX2, NOX3, NOX4, and p22phox. Specifically, the protein kinase RNA-like ER kinase (PERK) pathway is involved in the expression of NOX4 and p22phox, the inositol-requiring enzyme 1 alpha (IRE1α) pathway in NOX2 and NOX3 expression, and the activating transcription factor 6 (ATF6) pathway influences NOX3 expression in chondrocytes. Conversely, inhibiting NOX function significantly reduced both ER stress sensor–related signaling and chondrocyte catabolism, thereby decelerating the progression of surgically induced OA in vivo.
Conclusions
Our findings highlight the positive feedback loop between ER stress and oxidative stress in OA pathogenesis, suggesting that targeting NOX isoforms is a promising therapeutic strategy for OA.
Introduction
Chondrocytes appear to remain in a quiescent state with insufficient regenerative capacity within the dense, hypoxic extracellular matrix (ECM) environment. 1 However, they are highly active in producing a diverse range of ECM proteins, notably type II collagen (Col2) and aggrecan.1-3 These proteins are essential for providing structural support, tensile strength, and elasticity to the cartilage, thereby maintaining cartilage homeostasis and offering the necessary framework for proper joint function.2,3 Recent evidence has shown that chondrocytes from lesional cartilage exhibit accelerated protein synthesis compared with chondrocytes from non-lesional cartilage in the same osteoarthritis (OA) patient.3,4
The protein synthesis requires intricate processes, such as protein-folding and post-translational modifications, which take place in the endoplasmic reticulum (ER). 5 However, in the presence of cellular stress, the protein-folding capacity of the ER may become insufficient to meet the increased demand, resulting in an accumulation of misfolded or unfolded proteins within the ER. 6 This accumulation leads to ER stress, which subsequently triggers the activation of the unfolded protein response (UPR).6,7 UPR is mainly mediated by 3 ER transmembrane sensors, called protein kinase RNA-like ER Kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 α (IRE1α). 6 The primary goal of the UPR is to restore ER homeostasis by improving the protein-folding capacity, reducing protein synthesis, and facilitating the degradation of misfolded proteins. 7 However, when the ER stress persists, the UPR can have detrimental effects on chondrocytes, shifting the balance toward catabolic processes.7,8 Indeed, while mild ER stress can be protective for chondrocytes by activating autophagy via the 78-kDa glucose-regulated protein (GRP78) pathway, excessive ER stress can lead to chondrocyte apoptosis, ultimately contributing to the progression of OA.9,10 Therefore, chondrocytes have evolved mechanisms to counteract excessive ER stress. For instance, the AMP-activated protein kinase (AMPK) signaling can inhibit CHOP-mediated chondrocyte apoptosis, 11 and the sirtuin-1 attenuates the PERK-eIF-2α-CHOP axis by deacetylating PERK, thereby maintaining cartilage homeostasis. 12
The pathophysiologic stresses that can induce ER stress and the UPR include hyperglycemia, hyperlipidemia, hypoxic stress, oxidative stress, calcium imbalance, and nutrient deprivation.8,13 Among them, increased oxidative stress impairs the protein folding process and disulfide bond formation, ultimately leading to ER stress, which can be mitigated by antioxidants, like N-acetyl-cysteine.14,15 Not only does reactive oxygen species (ROS) induce ER stress, but recent evidence highlights that ER stress can in turn enhance ROS production. 16 This bidirectional relationship between ER stress and ROS forms a positive feedback loop that can amplify cellular stress responses and promote catabolic processes, synergistically contributing to cartilage degeneration in OA.17,18 However, there is a lack of research exploring the detailed mechanisms through which ER stress increases ROS production. In our previous study, we reported that the disruption of the ECM in a chondroitin sulfate-based 3D hydrogel increases oxidative stress in chondrocytes, potentially involving the NADPH oxidase (NOX) and mitochondrial ROS systems.19,20 These findings suggest that the breakdown of the cartilage ECM due to excessive mechanical stress may result in increased ROS production, leading to elevated ER stress in the context of OA.
In this study, we aimed to investigate the molecular mechanisms through which ER stress enhances ROS production in chondrocytes. We found that NOX system plays an important role in ROS production and catabolic response in chondrocytes under ER stress condition. Then, we investigated which ER stress sensor signaling pathways control the expression of NOX isoforms. Finally, we examined the role of inhibiting NOX isoforms both in the ER stress sensor signaling and catabolic response in chondrocytes, as well as in an in vivo, surgically induced OA model. By elucidating the molecular mechanisms underlying the crosstalk between ER stress and ROS in chondrocytes, we aim to provide new insights into the pathophysiology of OA and identify potential therapeutic targets for the treatment of this debilitating disease.
Materials and Methods
Primary Chondrocyte Cultures
All animal experiments were approved by the Animal Care Committee of Kyungpook National University (Approval No. KNU-2022-0203) and were performed in accordance with the guidelines of United States’ Public Health Service (PHS) Policy on Use of Laboratory Animals.
21
Primary chondrocytes were obtained from the articular cartilage of 5-day-old C57BL/6J mice by excising the distal femur and proximal tibia, which were then minced into small pieces. The cartilage pieces underwent a 15-minute incubation in 0.25% trypsin-EDTA at 37°C with gentle shaking and then were digested in 3 mg/mL of Collagenase P (Sigma-Aldrich) within complete Dulbecco’s Modified Essential Media (DMEM) for 4 hours. After digestion, the cell suspension was filtered through a 40-μm nylon mesh to eliminate undigested tissue. The chondrocytes were centrifuged at 300 x g for 5 minutes, washed twice with phosphate-buffered saline (PBS), and then resuspended in a 3:2 F12:DMEM-based culture medium with 10% fetal bovine serum (FBS), 0.25%
To investigate the effects of ER stress on primary chondrocytes, cells were treated with tunicamycin at concentrations of 0.5, 1.0, and 2 μg/mL or with equivalent volume of dimethyl sulfoxide (DMSO) (0.1% v/v) as control, and incubated for 6 hours in RNA isolation and 24 hours in protein isolation and staining. To investigate the cellular origin of ROS under ER stress conditions, cells were pre-treated with an ER stress inhibitor, tauroursodeoxycholic acid (TUDCA), and various ROS system inhibitors, including 2-deoxy-
Analysis of Oxidative Stress: Detection of Intracellular ROS and Oxidative DNA Damage
Intracellular and mitochondrial ROS production, as well as oxidative DNA damage in primary chondrocytes, were assessed using dihydroethidium (DHE) and mitoSOX Red (ThermoFisher Scientific, #M36008) fluorescent dyes for ROS, and the 8-oxo-dG antibody for DNA damage, respectively. Primary chondrocytes were fixed with 4% paraformaldehyde for 10 minutes, permeabilized using 0.25% Triton X-100, and then rinsed 3 times in PBS. To assess intracellular ROS, chondrocytes were incubated with 5 μM DHE for 30 minutes at 37°C, or incubated with 2.5 μM mitoSOX Red for 10 minutes at 37°C, and washed with PBS, and then counterstained with DAPI. Images were captured using a KI-3000F fluorescence microscope (Korealabtech, South Korea). For immunofluorescence staining of 8-oxo-dG, cells were incubated with the primary 8-oxo-dG antibody (sc-66036; Santa Cruz Technology, Beverly, MA) overnight at 4°C. Then, cells were incubated with a secondary antibody for 2 hours, counterstained with DAPI, and imaged using a fluorescence microscope. Fluorescence-positive cells were quantified using ImageJ software (version 1.8.0, National Institutes of Health, Bethesda, MD).
Quantitative Real-Time Reverse Transcription-PCR
From cultured primary chondrocytes, total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA), followed by the generation of the first-strand cDNA with Superscript III reverse transcriptase (Invitrogen). A ViiA 7 Real-Time PCR System (Applied Biosystems, CA) and SYBR Green Master Mix (Applied Biosystems) were employed for real-time qPCR. Primers for real-time qPCR can be found in Suppl. Table S1. Target gene expression levels were calculated using the 2−ΔΔCT method and normalized to the geometric mean of Gapdh, which served as an internal control. All quantitative real-time reverse transcription-PCR (qRT-PCR) analysis were carried out in triplicate, repeated 3 to 5 times, and the average results for each were presented.
Western Blot Analysis and Quantification
Proteins from primary chondrocyte cultured in 6-well plates were extracted using 300 μL RIPA buffer supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). Total cell lysates containing 10 to 20 μg of protein were subjected to 10% SDS-polyacrylamide gel electrophoresis and subsequently transferred onto polyvinylidene difluoride membranes (Immobilon-P; Millipore Corporation, Billerica, MA). Membranes were blocked using 5% skimmed milk in PBS with 0.25% Tween-20 (PBST) and incubated with the primary antibodies overnight at 4°C detailed in Suppl. Table S2. After washing with PBST, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 hours and then blots were developed using enhanced chemiluminescence Western blotting detection reagent (Thermo Fisher Scientific) and examined with the MicroChemi system (DNR Bio-imaging Systems). All analysis was biologically triplicated, and the Western blot band intensities were quantified using ImageJ software (version 1.8.0, NIH). Band intensities of target proteins were normalized to the β-actin band intensity of respective lanes.
Induction of Surgical OA Model and APX115 Treatment
Surgical OA was induced in 7 male 12-week-old C57BL/6 mice per group by destabilization of the medial meniscus (DMM) surgery under general anesthesia. All mice were purchased from Hana Biotech Co. (Pyeongtaek-si, South Korea) and were housed at a temperature of 23°C ± 1°C with a 12-hour light/dark cycle. They were kept under specific pathogen-free conditions in the animal facilities of Kyungpook National University Chilgok Hospital. The sample size was chosen based on our prior study that demonstrated statistically significant effect sizes. 20 Before starting the treatment, a total of 14 mice that underwent DMM surgery were randomly allocated into 1 of 2 groups. They were housed 2 per plastic cage, and the placement of the cages in the room was also randomized. The treatment group received intraperitoneal injections of APX-115 at a dosage of 30 mg/kg twice weekly, whereas the control group received an equivalent volume of DMSO in the same frequency. The dosage of APX-115 was based on a previous study that reported significant anti-inflammatory effects through the oral route in diabetic mice, as well as on our prior research.20,22 Eight weeks after surgery, the mice were euthanized via cervical dislocation, and knee joints were collected for histological examination, and all specimens were used for analysis. Treatments were administered by a separate technician who was aware of the groupings, but all assessments, measurements, and analyses on the mice were performed by researchers unaware of the mice’s treatment assignments. This study was conducted in accordance with ARRIVE guidelines 2.0, and the associated ARRIVE guideline checklist has also been offered in the Supplementary sections.
Safranin-O Staining and Immunohistochemistry of Mouse Knee Joint
Mouse knee joints were fixed in 4% paraformaldehyde for 24 hours, decalcified in 10% EDTA for 3 weeks, and then embedded in paraffin. The embedded blocks were sectioned at a thickness of 6 μm and stained with Safranin-O/Fast Green. Cartilage destruction in the total all 4 quadrants of joint (grade 0-24) was scored by 2 observers under blinded conditions using the Osteoarthritis Research Society International (OARSI) score system. 23 For immunohistochemical staining, the rehydrated sections underwent antigen retrieval in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) or by boiling for 15 minutes at temperature ranging from 95°C to 100°C. After blocking the sections with 2% bovine serum albumin (BSA) in PBS for 1 hour, they were incubated overnight at 4°C with primary antibodies for MMP13, CHOP, Grp78, or normal rabbit IgG diluted in 1% BSA (Suppl. Table S2). Subsequently, the sections were incubated with a biotinylated anti-rabbit secondary antibody (BA-1000; Sigma-Aldrich) and visualized with the Vector Red Alkaline Phosphatase Kit (AK-5200, Vector Labs, CA). Sections were mounted with anti-fade mounting solution (Vector Labs) and imaged and quantified under the KI-3000F fluorescence microscope. The immunostaining images of MMP13, CHOP, and Grp78 were quantified in the upper part of the tide mark of lateral tibial plateau.
Statistical Analysis
All data are presented herein as mean ± standard deviation (SD). Statistical analyses to compare the mean values of 2 groups were performed using the Mann-Whitney U test, a nonparametric test selected due to the small sample size and the lack of a normality assumption. A P-value of less than 0.05 was considered statistically significant. All statistical analyses were carried out using Prism software version 8.0 (GraphPad Software, La Jolla, CA).
Results
ER Stress Increases Oxidative Stress, Primarily via the NOX System
First, we established the tunicamycin-induced ER stress condition. Tunicamycin was treated at various dosages, and the expression of UPR marker proteins was assessed. Tunicamycin increased the protein levels of CHOP, XBP1s, and Grp78 in a dose-dependent manner, with the most significant effects observed at a concentration of 2 μg/mL (
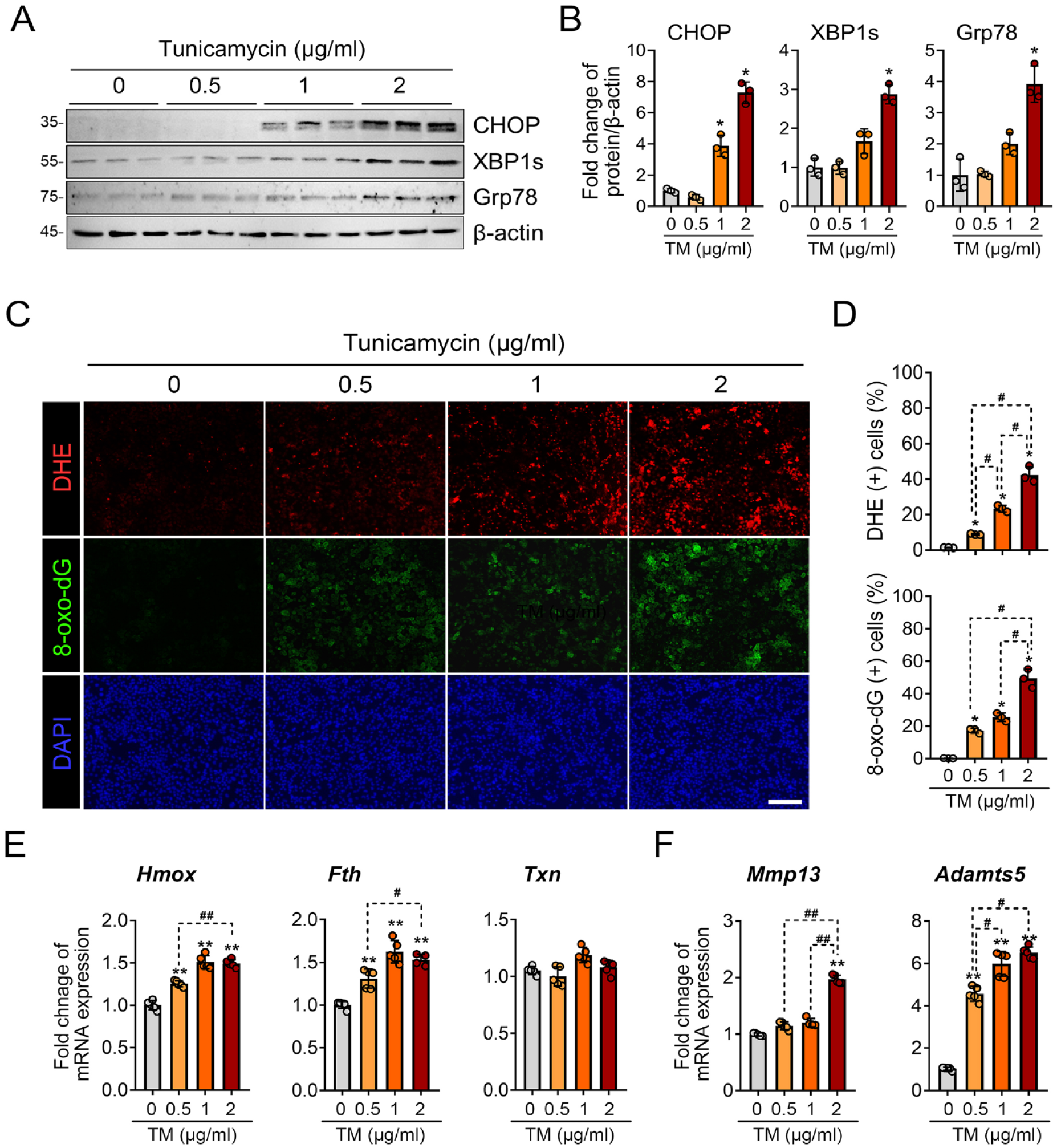
Tunicamycin-induced ER stress leads to oxidative stress in primary chondrocytes. (
We then explored the cellular origin of ROS under ER stress conditions in chondrocytes. For this, we used various inhibitors targeting different ROS systems, including TUDCA (an ER stress inhibitor), 2-deoxy-
ROS production under tunicamycin-induced ER stress is primarily dependent on the NOX and the mitochondrial electron transport chain system. (
ER Stress Upregulates the Expression of NOX2, NOX3, NOX4, and p22phox, but Not NOX1
Based on the findings of NOX-mediated ROS production under ER stress conditions, we investigated the expression patterns of different NOX isoforms in primary chondrocytes. ER stress induced by treatment with 2 μg/mL of tunicamycin led to the upregulation of RNA levels for NOX2 (gp91phox), NOX3, NOX4, and p22phox, a shared subunit of NOX1-4. However, the expression of NOX1 remained unaffected (
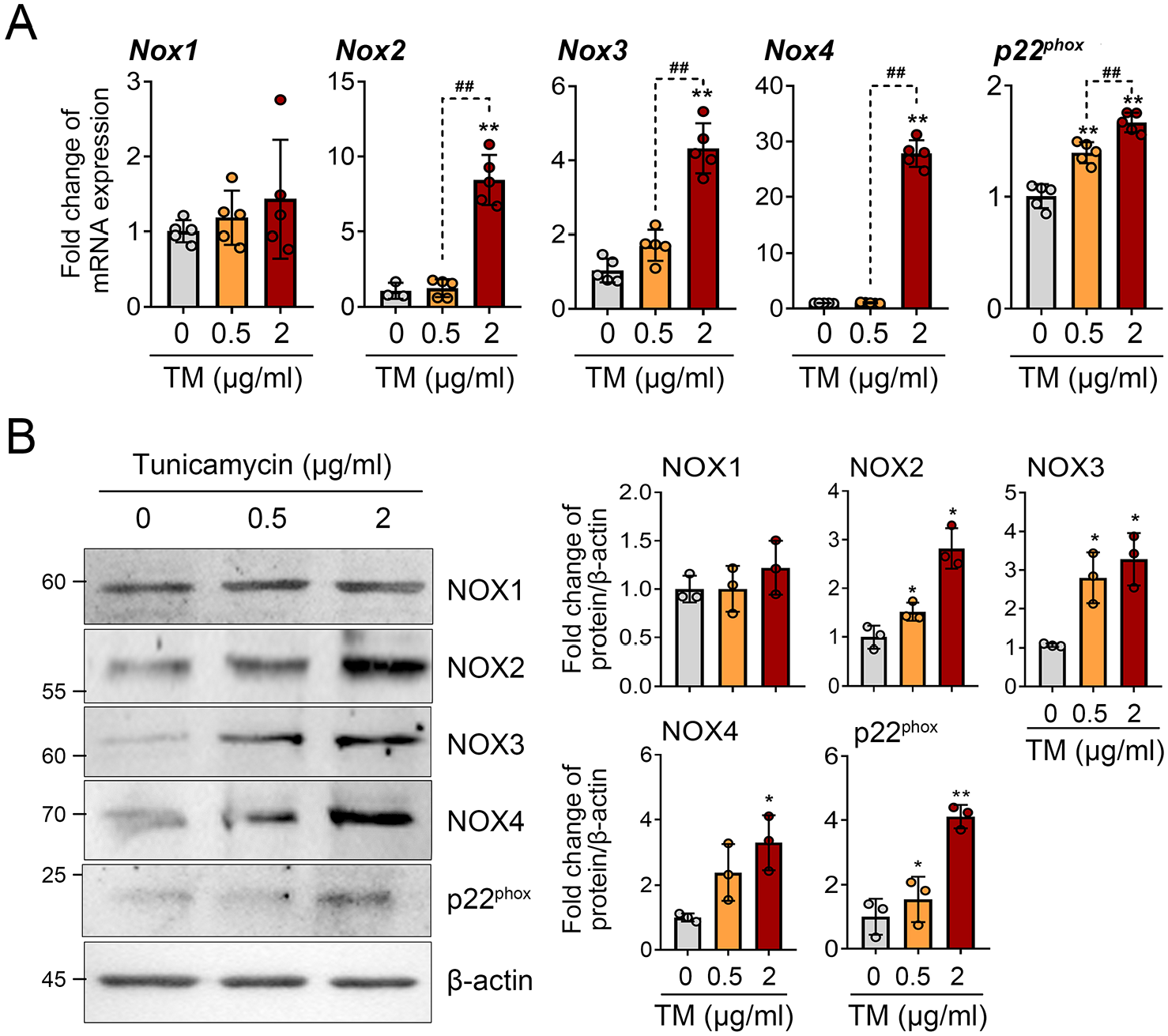
Tunicamycin-induced ER stress increases the expression of NOX2, NOX3, NOX4, and p22phox, but not NOX1. (
The PERK Pathway Upregulates NOX4 and p22phox, while the IRE1α Pathway Upregulates NOX2 and NOX3 under ER Stress Conditions
The 3 main ER stress sensor signaling pathways—PERK, IRE1α, and ATF6—are activated under ER stress conditions. We investigated which of these pathways regulate the increase of NOX isoforms under ER stress conditions. Upon treating the cells with the IRE1α inhibitor MKC 8866 and the PERK inhibitor GSK2606414, we observed significant reduction of their respective target proteins, XBP1s for the IRE1α pathway and CHOP for the PERK pathway in Western blot analysis. The 78-kDa glucose-regulated protein (GRP78) expression, known to be regulated by the ATF6 pathway,
24
was significantly suppressed by both ceapins and GSK2606414 (
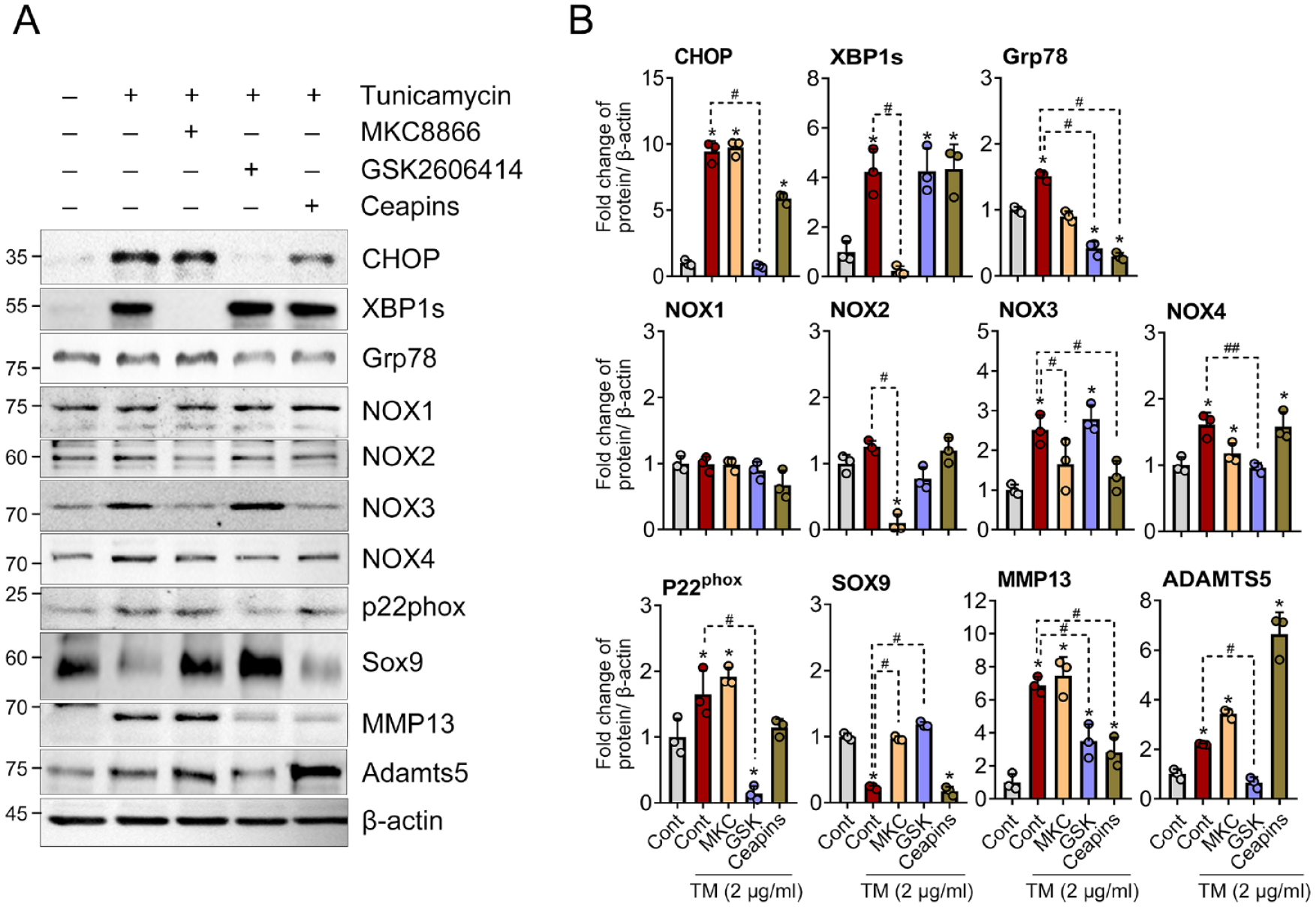
ER stress sensor signaling pathways regulate the expression of NOX isoforms and p22phox. (
NOX Inhibition Significantly Suppresses ER Stress Sensor Signaling and Chondrocyte Catabolism under ER Stress Condition
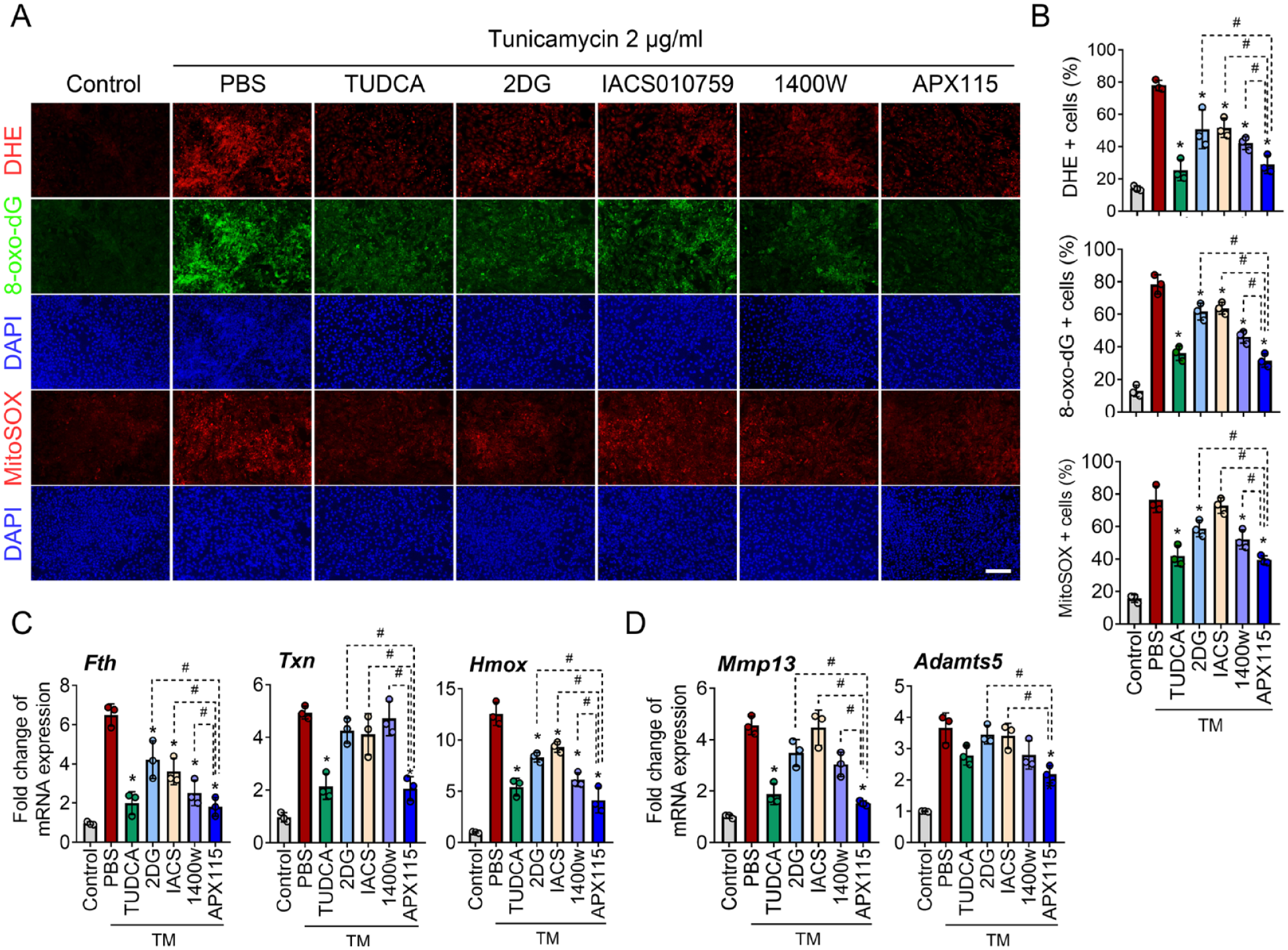
To determine the effect of NOX isoforms on oxidative stress under ER stress condition, we inhibited NOX isoforms using various inhibitors, including ML171 (NOX1 inhibitor), GSK2795039 (NOX2 inhibitor), GLX351322 (NOX4 inhibitor), GKT137831 (NOX1/4 inhibitor), and APX115 (pan-NOX inhibitor). When assessing ROS production using DHE, the pan-NOX inhibitor APX115 most effectively suppressed ROS production by tunicamycin, while inhibitors targeting NOX1, NOX2, NOX4, and NOX1/4 yielded similar, albeit lesser, levels of suppression. The expression of 8-oxo-dG was most profoundly suppressed by GSK2795039, a NOX2 inhibitor, followed by the NOX4 and pan-NOX inhibitors, both of which showed similar suppression levels of 8-oxo-dG (
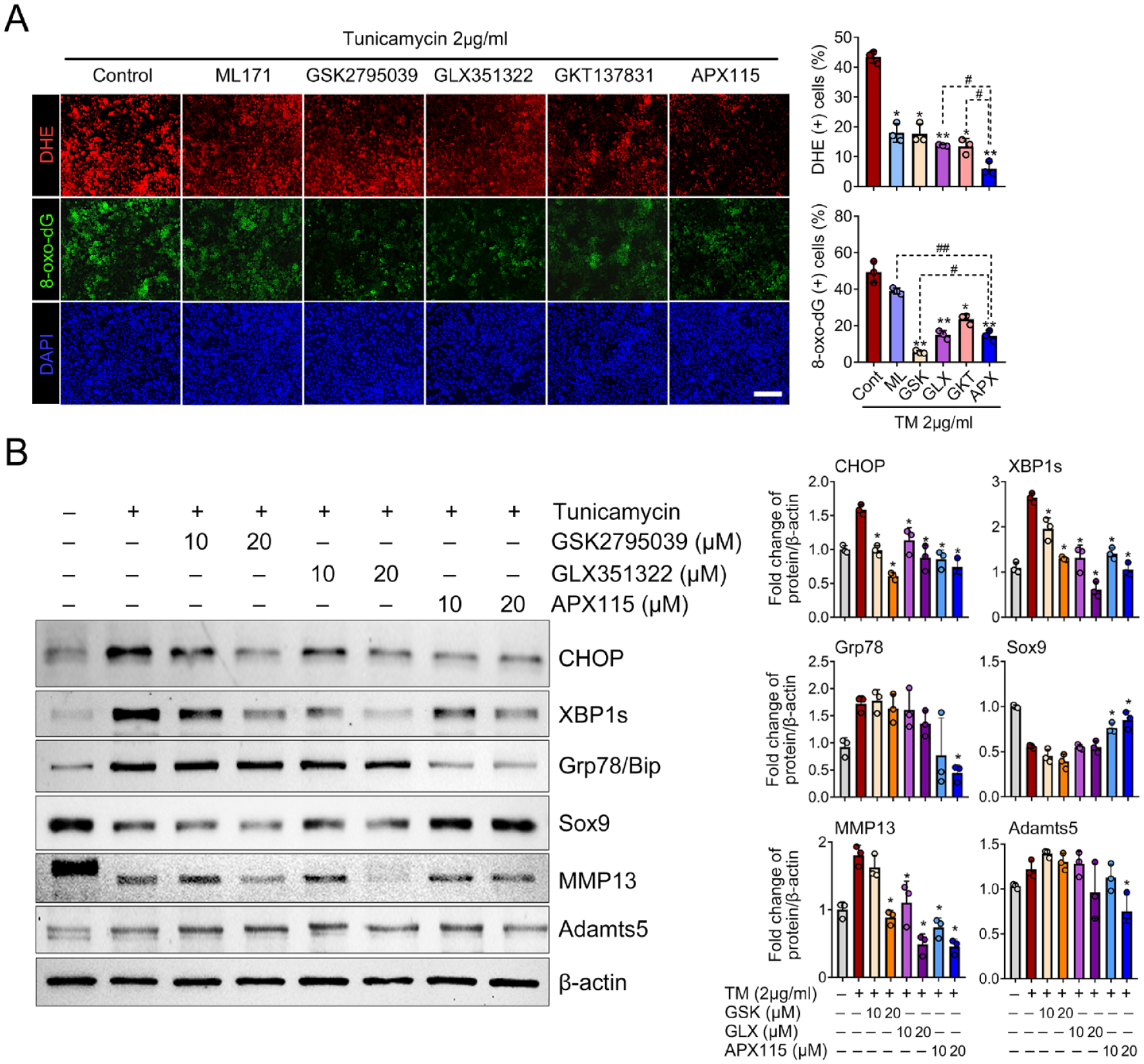
NOX-mediated oxidative stress is pivotal for ER stress sensor signaling. (
We further examined the effects of NOX inhibition on the activation of ER stress sensor signaling and chondrocyte catabolism in the context of tunicamycin-induced ER stress. Treatment with the NOX2 inhibitor GSK2795039, NOX4 inhibitor GLX351322, and the pan-NOX inhibitor APX115 significantly suppressed the expression of both CHOP and XBP1s. Grp78 expression was significantly suppressed only by a higher dose (20 μM) of the pan-NOX inhibitor, but not by NOX2 or NOX4 inhibitor alone. Moreover, the pan-NOX inhibitor led to a significant increase in Sox9 expression sufficiently to counteract the decrease by interleukin (IL)-1β, but NOX2 or NOX4 inhibitor failed to increase Sox9. The IL-1β-mediated increase of MMP13 was significantly suppressed by NOX2, NOX4, and pan-NOX inhibitor. In addition, Adamts5 was significantly reduced when treated with 20 µM APX115, whereas NOX2 or NOX4 inhibitor alone failed to induce a notable change (
NOX Inhibition Attenuates the Progression of Surgically Induced OA and Reduces the Expression of ER Stress Markers in Articular Cartilage
To prove the role of NOX-mediated oxidative stress in ER stress and its catabolic function, APX115, a pan-NOX inhibitor, was administered intraperitoneally to DMM-induced mice OA model. APX115 treatment significantly reduced both the total and tibial plateau OARSI grade, as well as the subchondral bone thickness, compared with the DMM controls (
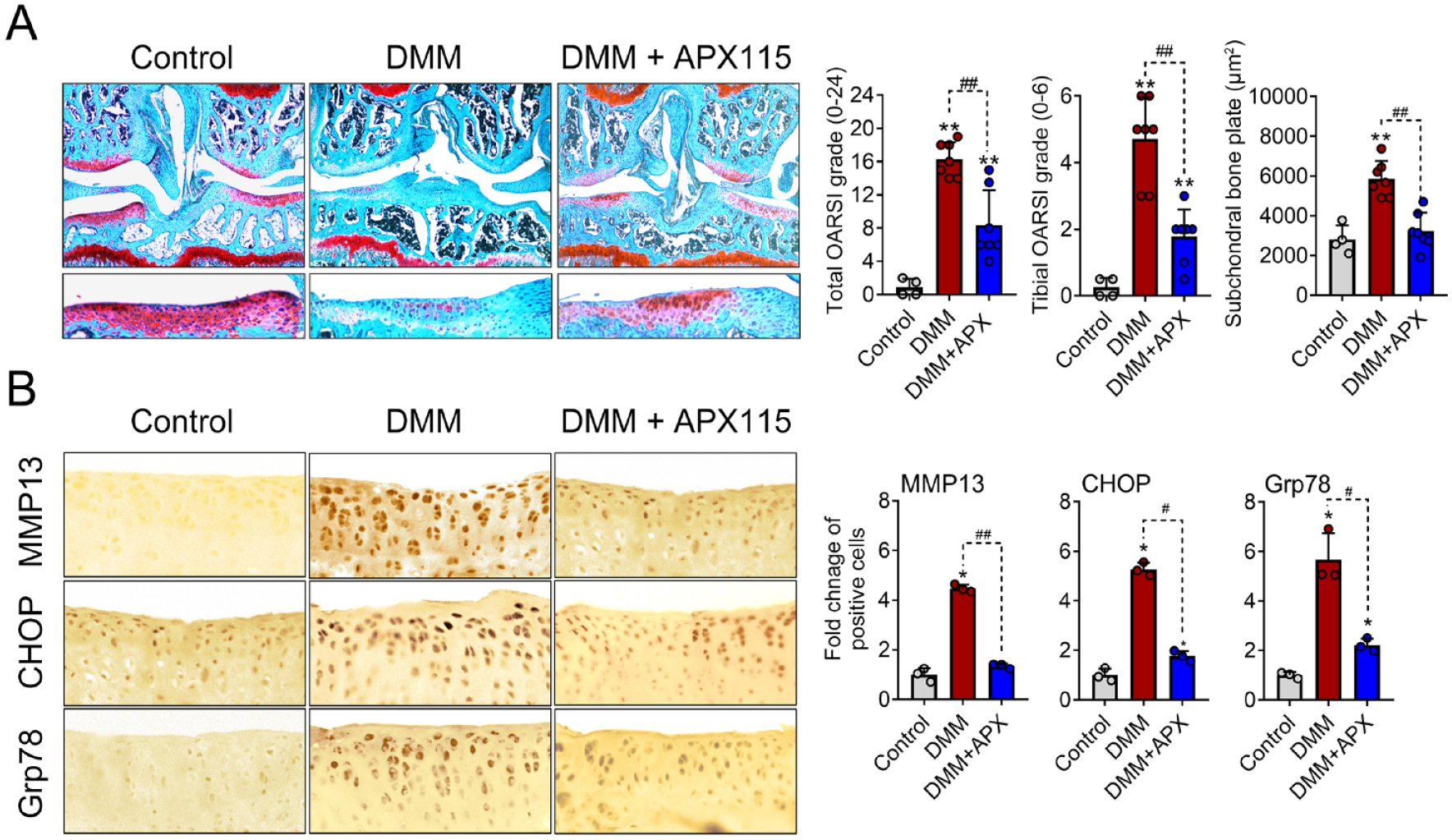
Inhibition of NOX-mediated oxidative stress not only prevents ER stress in the articular cartilage but also halts the progression of surgically induced OA. (
Discussion
Gene signatures linked to increased oxidative stress and ER stress are frequently observed in the cartilage tissue of OA patients.25,26 This study confirmed the importance of NOX system in the increase of ROS associated with ER stress. Each pathway of the ER stress sensor signaling specifically acted on the increase of NOX isoforms. In particular, the IRE1α pathway primarily influenced the upregulation of NOX2 and NOX3, whereas the PERK pathway was crucial for augmenting NOX4 and p22phox. The increase of the NOX system and the subsequent rise in ROS levels were pivotal in the ER stress response. Inhibiting NOX in vivo in the DMM model resulted in a marked reduction of ER stress markers in articular cartilage, thereby demonstrating a protective effect against the progression of OA.
We found that the ROS production is upregulated in chondrocytes under ER stress conditions induced by tunicamycin, which is primarily mediated by the NOX system (
ER stress can trigger 3 distinct signaling pathways, namely PERK, IRE1α, and ATF6 pathways. These pathways sense an accumulation of unfolded proteins in the ER lumen and relay signals to the cytosol, which are then directed toward the nucleus. While ATF6 directly functions as a transcription factor, the PERK and IRE1α pathways use downstream transcription factors like ATF4 and XBP1, respectively.
29
Most previous studies have focused on how NOX-mediated ROS amplifies ER stress and promotes catabolic responses, such as apoptosis.30-33 In this study, we aimed to elucidate the interaction between ER stress and the NOX system. Our most notable finding provides a detailed understanding of molecular mechanisms by which tunicamycin-induced ER stress controls the expression of NOX isoforms in chondrocytes. Specifically, NOX2 is regulated through the IRE1α pathway, NOX3 through both the IRE1α and ATF6 pathways, and NOX4 and p22phox through the PERK pathway (
The pivotal finding of our study highlights a positive feedback loop between the NOX system and ER stress signaling. The inhibition of NOX2 and NOX4 suppresses the 2 major ER stress sensor pathways of PERK, and IRE1α, and pan-NOX inhibition suppresses all 3 major ER stress pathways (
Our data showed that NOX2, 3, and 4 exhibited a dose-dependent increase in response to tunicamycin, while NOX1 maintained its expression without any increase in the presence of ER stress in chondrocytes (
NOX3 has been identified primarily in the vestibular and cochlear epithelia of inner ear, and the mice deficient in NOX3 exhibit a head-tilt phenotype. 38 However, the expression and role of NOX3 in tissues other than the inner ear are not well known. The datasheet for the commercial NOX3 antibody indicates a positive band in the mouse brain, pancreas, kidney, and liver tissues (novusbio, nox3-antibody_nbp2-94866). Our previous data revealed that primary chondrocyte from mice expresses NOX3, which is decreased by IL-1β treatment and OA articular cartilage. 20 Under ER stress condition examined in this study, NOX3 showed an increasing trend, which contrasts with catabolic situations such as OA or IL-1β treatment. 20 The function of NOX3 in cartilage physiology is still not well defined, and it warrants further in-depth research.
In conclusion, our study elucidates the interplay between ER stress and NOX-mediated oxidative stress in chondrocytes. The increase in unfolded proteins initiates ER stress, which activates 3 sensor signaling pathways, including PERK, IRE1α, and ATF6. Specifically, PERK signaling drives the expression of NOX4 and p22phox, IRE1α controls NOX2 expression, and both IRE1α and ATF6 jointly modulate NOX3. This enhanced expression of NOX isoforms further exacerbates ER stress via the amplification of oxidative stress (
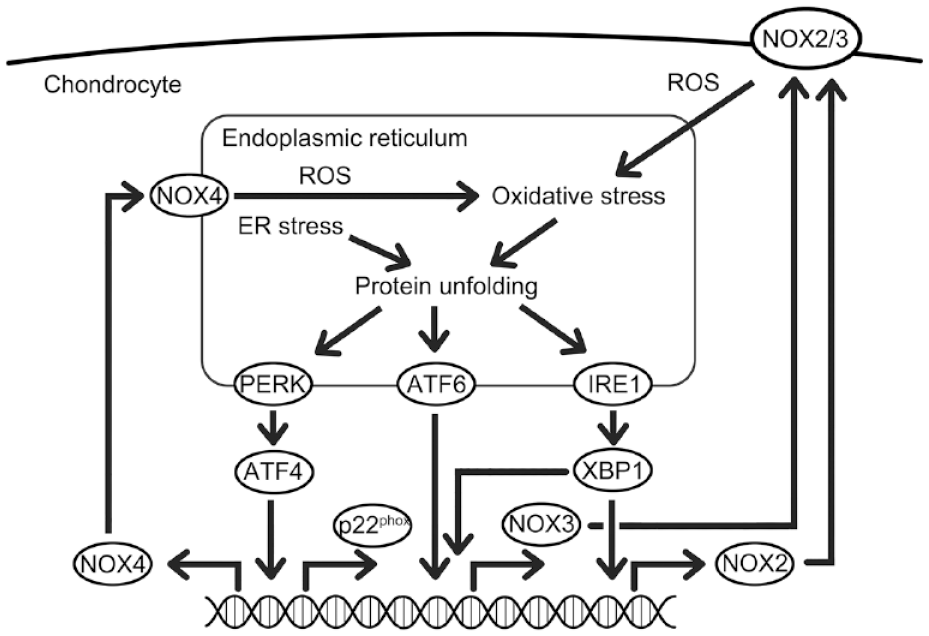
A summary of this study showing the interplay between ER stress and NOX-mediated oxidative stress in chondrocyte catabolism. ER stress by the unfolded protein response triggers 3 ER stress sensor signaling pathways. These pathways increase NOX isoforms: PERK affects NOX4 and p22phox, IRE1α affects NOX2 and NOX3, and ATF6 targets NOX3. The NOX-mediated oxidative stress, in turn, boosts ER stress, creating a feedback loop that might contribute to the deterioration of OA cartilage.
Supplemental Material
sj-pdf-1-car-10.1177_19476035241245803 – Supplemental material for The Interplay Between Endoplasmic Reticulum Stress and Oxidative Stress in Chondrocyte Catabolism
Supplemental material, sj-pdf-1-car-10.1177_19476035241245803 for The Interplay Between Endoplasmic Reticulum Stress and Oxidative Stress in Chondrocyte Catabolism by Yu Jung Kim, Jin Han and Seungwoo Han in CARTILAGE
Supplemental Material
sj-pdf-2-car-10.1177_19476035241245803 – Supplemental material for The Interplay Between Endoplasmic Reticulum Stress and Oxidative Stress in Chondrocyte Catabolism
Supplemental material, sj-pdf-2-car-10.1177_19476035241245803 for The Interplay Between Endoplasmic Reticulum Stress and Oxidative Stress in Chondrocyte Catabolism by Yu Jung Kim, Jin Han and Seungwoo Han in CARTILAGE
Footnotes
Author Contributions
Study conception and design: Y.J.K. and S.H. Acquisition of data: Y.J.K. and J.H. Analysis and/or interpretation of data: Y.J.K. and S.H. Wrote the paper: Y.J.K. and S.H. Critical revision: Y.J.K., J.H. and S.H. All authors read and approved the final manuscript.
Acknowledgments and Funding
We would like to express our sincere gratitude to Professor Ji-young Park, Pathology of Kyungpook National University, for her invaluable guidance, expertise, and unwavering support throughout this research. This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (grant number RS-2022-00243140).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval
All experiments were conducted in accordance with approved animal protocols and guidelines established by the Animal Care Committee of Kyungpook National University (Approval No. KNU-2018-62/54).
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.