
Abstract
Objective
Osteoarthritis (OA) is characterized by progressive cartilage degeneration driven by inflammation-induced chondrocyte injury, while effective disease-modifying therapies are still lacking. Exosome-based cell-free approaches are emerging as promising alternatives, but their key molecular mediators remain incompletely defined.
Design
Adipose-derived stem cells (ADSCs) were pretreated with platelet-rich plasma (PRP) to enhance exosomal function. Isolated exosomes were characterized, and LINC01106 expression was modulated by overexpression or knockdown. Interleukin (IL)-1β-stimulated chondrocytes were used to assess apoptosis, inflammatory cytokine secretion, oxidative stress, and extracellular matrix degradation. The LINC01106/miR-34a-5p/SIRT1 axis was examined using luciferase reporter assays, quantitative real-time polymerase chain reaction (PCR), Western blotting, and immunofluorescence.
Results
PRP pretreatment markedly increased LINC01106 enrichment in ADSC-derived exosomes. LINC01106-rich exosomes significantly reduced chondrocyte apoptosis, suppressed tumor necrosis factor-α (TNF-α) and IL-6 secretion, alleviated oxidative stress, and attenuated matrix metalloproteinase (MMP)-mediated matrix degradation under IL-1β stimulation. Mechanistically, LINC01106 acted as a competing endogenous RNA that sequestered miR-34a-5p, thereby restoring SIRT1 expression. Rescue experiments demonstrated that miR-34a-5p overexpression or SIRT1 silencing abolished these protective effects. In addition, LINC01106-enriched exosomes inhibited nuclear factor-κB (NF-κB) and mitogen-activated protein kinase (MAPK) pathway activation in a SIRT1-dependent manner.
Conclusion
PRP-stimulated ADSC-derived exosomes confer potent chondroprotective effects through the LINC01106/miR-34a-5p/SIRT1 pathway, highlighting a promising cell-free therapeutic strategy for OA.
Introduction
Osteoarthritis (OA) is a long-standing degenerative disorder of the joints, marked by progressive cartilage erosion and ongoing synovial inflammation, and represents a leading contributor to global disability and diminished life quality.1,2 A sustained low-grade inflammatory microenvironment within the joint cavity is increasingly recognized as central to OA pathogenesis.3,4 Interleukin-1β (IL-1β), which is substantially increased in both synovial fluid and cartilage tissues of patients with OA, 5 drives chondrocyte dysfunction by inducing matrix-degrading enzymes, including matrix metalloproteinases (MMPs) and aggrecanases, thereby accelerating extracellular matrix breakdown and promoting apoptosis and senescence, ultimately leading to irreversible cartilage damage.6,7 Accordingly, suppressing IL-1β–mediated inflammatory injury in chondrocytes is a key strategy to slow OA progression.
Adipose-derived mesenchymal stem cells (ADSCs) are gaining growing attention as a potential therapeutic option for OA, owing to their easy accessibility, minimal immunogenic response, and potent paracrine signaling capacity.8,9 However, direct stem cell transplantation is limited by poor cell survival and safety concerns. 10 Consequently, ADSC-derived exosomes have gained attention as a cell-free alternative, mediating intercellular communication through the delivery of bioactive cargos while avoiding potential complications associated with live-cell therapies.11,12 Moreover, preconditioning can further enhance exosome efficacy. As a growth factor–enriched autologous product, platelet-rich plasma (PRP) can stimulate ADSC activity, leading to increased exosome production and augmented biological effects, which may serve as an effective means to optimize exosome-centered interventions for OA.13,14
Mechanistically, exosomal effects are largely attributed to functional non-coding RNAs, particularly long non-coding RNAs (lncRNAs) and microRNAs (miRNAs), which orchestrate post-transcriptional regulation of gene expression. 15 LncRNAs can act as competing endogenous RNAs (ceRNAs) by sequestering specific miRNAs and thereby relieving miRNA-mediated repression of downstream target messenger RNAs (mRNAs). Dysregulated ceRNA networks have been increasingly implicated in OA initiation and progression, representing a key layer of post-transcriptional regulation in joint disease.16,17 LINC01106 (Long Intergenic Non-Protein Coding RNA 1106) is a recently characterized lncRNA that has been shown to be aberrantly expressed across multiple pathological contexts. In oncological settings, LINC01106 functions as an oncogenic driver by promoting tumor cell proliferation, migration, and invasion in colorectal cancer, gastric cancer, and lung adenocarcinoma, in part through its capacity to act as a ceRNA that sequesters tumor-suppressive miRNAs.18,19 Notably, LINC01106 has been demonstrated to directly sponge miR-34a-5p, thereby relieving its suppression of downstream targets. 18 Beyond cancer, emerging transcriptomic evidence from musculoskeletal diseases has identified LINC01106 as a differentially expressed lncRNA in pathological bone and joint conditions, where it participates in ceRNA netwo rks involving miR-34a-5p. 20 However, the functional role of LINC01106 in OA chondrocytes and its potential to modulate inflammatory injury through miRNA-mediated mechanisms remain to be elucidated.
Sirtuin 1 (SIRT1) is a class III histone deacetylase that requires nicotinamide adenine dinucleotide (NAD⁺) for activity and serves as an essential regulator of cellular equilibrium, inflammatory responses, and aging processes. 21 In chondrocytes, SIRT1 suppresses inflammation by deacetylating the p65 subunit of nuclear factor-κB (NF-κB), resulting in reduced NF-κB activity and limiting pro-inflammatory mediator expression. 22 In contrast, miR-34a-5p is a critical pro-inflammatory miRNA in OA that promotes apoptosis and extracellular matrix degradation and correlates with disease severity, suggesting that miRNA-centered axes converging on SIRT1/NF-κB are key nodes in inflammatory cartilage injury. 23
Although PRP-preconditioned ADSC-derived exosomes have shown therapeutic potential in OA, whether exosome-enriched lncRNAs modulate miRNA activity and downstream inflammatory signaling through ceRNA-based mechanisms remains insufficiently defined. Mechanistic insights into OA therapy may be advanced by analyzing lncRNA-mediated ceRNA networks in exosomes derived from PRP-conditioned ADSCs. The investigation assesses how such networks regulate inflammatory responses and matrix breakdown in chondrocytes under IL-1β stimulation, thereby informing the design of cell-free exosome therapeutic strategies. Therefore, the present study aimed to investigate whether LINC01106, an lncRNA enriched in PRP-stimulated ADSC-derived exosomes, exerts chondroprotective effects in IL-1β–induced OA chondrocytes. We hypothesized that exosomal LINC01106 alleviates chondrocyte inflammatory injury by functioning as a ceRNA to sequester miR-34a-5p, thereby restoring SIRT1 expression and suppressing downstream NF-κB and mitogen-activated protein kinase (MAPK) inflammatory signaling.
Materials and Methods
Study Design
This was an in vitro, comparative, mechanistic study. The study comprised 3 main experimental groups: (1) untreated C28/I2 chondrocytes (negative control), (2) IL-1β–stimulated C28/I2 chondrocytes (OA model/positive control), and (3) IL-1β–stimulated C28/I2 chondrocytes treated with ADSC-derived exosomes with varying LINC01106 expression levels (experimental groups). The primary outcomes included chondrocyte viability, apoptosis rate, inflammatory cytokine secretion (tumor necrosis factor-α (TNF-α), IL-6), oxidative stress markers (superoxide dismutase (SOD), malondialdehyde (MDA)), and extracellular matrix component expression (MMP-13, COL2A1, ACAN, COMP). Using the PICO framework: the Population was IL-1β–stimulated C28/I2 chondrocytes; the Intervention was treatment with LINC01106-enriched ADSC-derived exosomes; the Comparison was untreated or LINC01106-depleted exosome groups; and the Outcomes were the above-mentioned cellular and molecular endpoints.
Cell Culture and OA Model Construction
ADSCs and the human immortalized chondrocyte cell line C28/I2 were obtained from a commercial cell bank. Culture of ADSCs was conducted using Dulbecco’s Modified Eagle Medium (DMEM), enriched with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P/S). C28/I2 chondrocytes were propagated in DMEM/F12 medium containing 10% FBS, 1% P/S, and 1% ITS (insulin-transferrin-selenium). Cells were used between passages 3 and 8 to ensure consistent phenotypic stability, in accordance with the manufacturer’s guidelines indicating that C28/I2 cells maintain stable chondrocyte marker expression for at least 10 passages after initial thawing. Incubation for all cell types occurred at 37°C within a humidified 5% CO₂ environment.
For the construction of an in vitro OA model, C28/I2 chondrocytes were seeded into six-well plates at a density of 3 × 10⁵ cells/well and allowed to adhere overnight prior to treatment. Cells were then stimulated with recombinant human IL-1β at concentrations of 10, 20, or 40 ng/ml for 24 h. These concentrations were selected based on previous studies demonstrating dose-dependent inflammatory responses in chondrocytes, 24 and 10 ng/ml was determined as the optimal concentration for subsequent experiments based on assessments of inflammatory responses and cell viability. It is acknowledged that these concentrations exceed the physiological levels of IL-1β typically detected in OA synovial fluid (generally in the range of 0.1-0.3 ng/ml in vivo). 25 However, in vitro systems lack the complex tissue architecture and cellular buffering mechanisms present in vivo, necessitating higher exogenous cytokine concentrations to achieve comparable intracellular signaling responses. The concentrations used in the present study are consistent with those employed in numerous published in vitro OA models,26,27 and the 10 ng/ml dose was selected as the minimum effective concentration based on dose-response assessments of cell viability and inflammatory markers.
Isolation and Characterization of Exosomes Derived From PRP-Treated ADSCs
PRP was prepared by 2-step centrifugation of freshly collected blood: an initial spin at 200 ×g for 10 min to remove red blood cells, followed by a second spin at 1,000 ×g for 10 min to concentrate platelets. The platelet concentration in the final PRP preparation was 1,171.80 ± 384.46 × 10⁹/l, representing approximately 5.2-fold enrichment over baseline whole blood. ADSCs were divided into 2 experimental conditions: untreated controls and PRP-stimulated cells. The PRP group was incubated for 48 h in exosome-depleted culture medium containing 5% PRP, whereas control cells were maintained in exosome-free medium alone. 28 Cell culture supernatants were obtained and fractionated by differential centrifugation for extracellular vesicle extraction. Briefly, sequential centrifugation steps included 300 ×g (10 min), 2,000 ×g (20 min), and 10,000 ×g (30 min) to remove contaminants, after which exosomes were pelleted by ultracentrifugation at 100,000 ×g for 70 min. The pellets were washed and resuspended in phosphate-buffered saline (PBS).
Nanoparticle tracking analysis (NTA) was applied to determine exosome size distribution and concentration. Confirmation of exosomal purity was achieved through Western blotting, which identified the presence of positive markers (CD63 and TSG101) and the absence of the negative marker calnexin.
For co-culture experiments, C28/I2 chondrocytes were seeded into six-well plates at a density of 3 × 10⁵ cells/well and allowed to adhere overnight. Following IL-1β stimulation (10 ng/ml, 24 h) to establish the in vitro OA model, the medium was replaced and cells were treated with PRP-ADSC-derived exosomes at a concentration of 80 μg/ml for 24 h. The exosome concentration was determined based on total protein content as measured by bicinchoninic acid (BCA) assay, consistent with concentrations reported in comparable in vitro studies of mesenchymal stem cell (MSC)-derived exosomes in chondrocyte inflammatory models. 29 Control groups received an equivalent volume of PBS.
Plasmid Construction and Cell Transfection
To modulate LINC01106 and miR-34a-5p expression, corresponding plasmids and RNA oligonucleotides were synthesized by a commercial provider. Constructs included a LINC01106 overexpression plasmid (pLVX-LINC01106), LINC01106-specific siRNA (si-LINC01106), and their respective negative controls (pLVX-NC and si-NC). Similarly, miR-34a-5p mimics and inhibitors, together with matched control sequences (mimic-NC and inhibitor-NC), were obtained commercially. For transfection, Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) was utilized according to the standard instructions.
Cell Proliferation and Apoptosis Assays
Cell growth was evaluated using the Cell Counting Kit-8 (CCK-8). After seeding cells into 96-well plates (5 × 10³ cells/well) and applying the designated treatments, each well received 10 μL of CCK-8 reagent and was incubated for 2 h. Finally, a microplate reader measured absorbance at 450 nm.
Apoptosis was evaluated using Annexin V-FITC/PI dual staining. Treated cells were harvested, subjected to 2 washes with chilled PBS, and then suspended in binding buffer. Annexin V-FITC and propidium iodide (PI) were added sequentially, and samples were incubated in the dark for 15 min at room temperature before immediate analysis by flow cytometry.
Enzyme-Linked Immunosorbent Assay
Inflammatory cytokine production was assessed by collecting cell culture supernatants, followed by enzyme-linked immunosorbent assay (ELISA)-based quantification of TNF-α (ml106471; mlbio, Shanghai, China) and IL-6 (KE00385; Proteintech, Rosemont, IL, USA) using manufacturer-recommended kits. In addition, cell lysates were harvested to assess oxidative stress markers, including SOD (ml063052; mlbio) and MDA (ml950271; mlbio).
Quantitative Real-Time Polymerase Chain Reaction
RNA extraction from all treatment groups utilized TRIzol reagent (Invitrogen). Reverse transcription for miRNA detection employed a miRNA-specific cDNA synthesis kit (Takara, Kusatsu, Shiga, Japan), while mRNA and lncRNA detection used PrimeScript RT Master Mix (Takara) for cDNA synthesis. Quantitative PCR reactions were run on a real-time PCR system with SYBR Green Master Mix (Takara). Normalization of miRNA data involved U6 as an internal control, and GAPDH was used to normalize mRNA and lncRNA expression. Expression levels were relatively quantified through the 2−ΔΔCt approach.
30
The sequences of all primers are summarized in
PCR Primer Sequences Applied in This Study.

Western Blot Analysis
Cells were disrupted using RIPA lysis buffer (Beyotime, Haimen, China). Protein content was quantified using a BCA assay (Beyotime). Equal protein loads were separated via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes. Membranes were blocked using 5% skim milk and subsequently exposed to primary antibodies at 4°C overnight against COMP (28369-1-AP, 1:1000; Proteintech), NF-κB p65 (#8242, 1:1000; CST, Danvers, MA, USA), MAPK (#9212, 1:1000; CST), COL2A1 (ab188570, 1:5000; Abcam), ACAN (#28971, 1:1000; CST), SIRT1 (#9172, 1:1000; CST), MMP-13 (ab39012, 1:3000; Abcam Cambridge, UK), and GAPDH (10494-1-AP, 1:5000; Proteintech) as an internal control. After thorough washing, membranes were probed with HRP-linked Goat Anti-Rabbit secondary antibodies (SA00001-2, 1:2000; Proteintech) at ambient temperature for 1 h. Detection was carried out with a chemiluminescent ECL substrate (Millipore, Burlington, MA, USA), and densitometry of immunoreactive bands was measured via ImageJ.
Immunofluorescence Staining
Cells grown on sterile coverslips and subjected to designated treatments. After fixation with 4% paraformaldehyde and permeabilization using 0.5% Triton X-100, nonspecific interactions were prevented by incubation with 5% bovine serum albumin (BSA). Primary antibodies targeting SIRT1 (#8469, 1:100; CST), NF-κB p65 (#8242, 1:100; CST), or MAPK (#9212, 1:1000; CST). The following day, exposed to appropriate fluorescent secondary antibodies for 60 min at ambient temperature while protected from light. Nuclei were visualized by DAPI, and images were recorded using a fluorescence imaging system.
Dual-Luciferase Reporter Assay
The potential direct regulatory interaction involving LINC01106, miR-34a-5p, and the SIRT1 3′UTR was tested using a dual-luciferase reporter system. Predicted miR-34a-5p binding regions within LINC01106 wild-type (WT) or mutant (MUT) and the SIRT1 3′UTR (WT or MUT) were individually cloned into the pmirGLO vector. 31
In C28/I2 cells, the prepared reporter plasmids were introduced together with miR-34a-5p mimics or mimic-NC. Luciferase activity was quantified 48 h later with a commercial dual-luciferase detection kit. Normalized relative reporter activity was derived by normalizing firefly luciferase signals to Renilla luciferase measurements.
Statistical Analysis
Experimental results are expressed as mean ± standard deviation (SD), with all assays performed in a minimum of 3 independent replicates. Data analysis was carried out using GraphPad Prism 9.5. For comparisons involving 2 groups, Student’s t-test was utilized; differences among multiple groups were assessed via 1-way analysis of variance (ANOVA) and subsequent Tukey’s post hoc test. Statistical significance was defined as P < 0.05. Prior to parametric testing, data normality was assessed using the Shapiro-Wilk test, and variance homogeneity was verified by Levene’s test. All experiments were performed with a minimum of 3 biological replicates (independent cell passages), each conducted in triplicate as technical replicates. The exact sample size for each experiment is indicated in the corresponding figure legends. Effect sizes were estimated using Cohen’s d for pairwise comparisons and partial η² for ANOVA analyses.
Results
IL-1β Induces Inflammatory Damage in C28/I2 Chondrocytes and Defines an OA-Like In Vitro Model
To mimic OA-associated inflammatory injury in vitro, C28/I2 chondrocytes were challenged with graded concentrations of IL-1β (10, 20, and 40 ng/ml). CCK-8 assays showed that IL-1β exposure led to a clear, concentration-dependent decline in chondrocyte viability compared with the untreated control
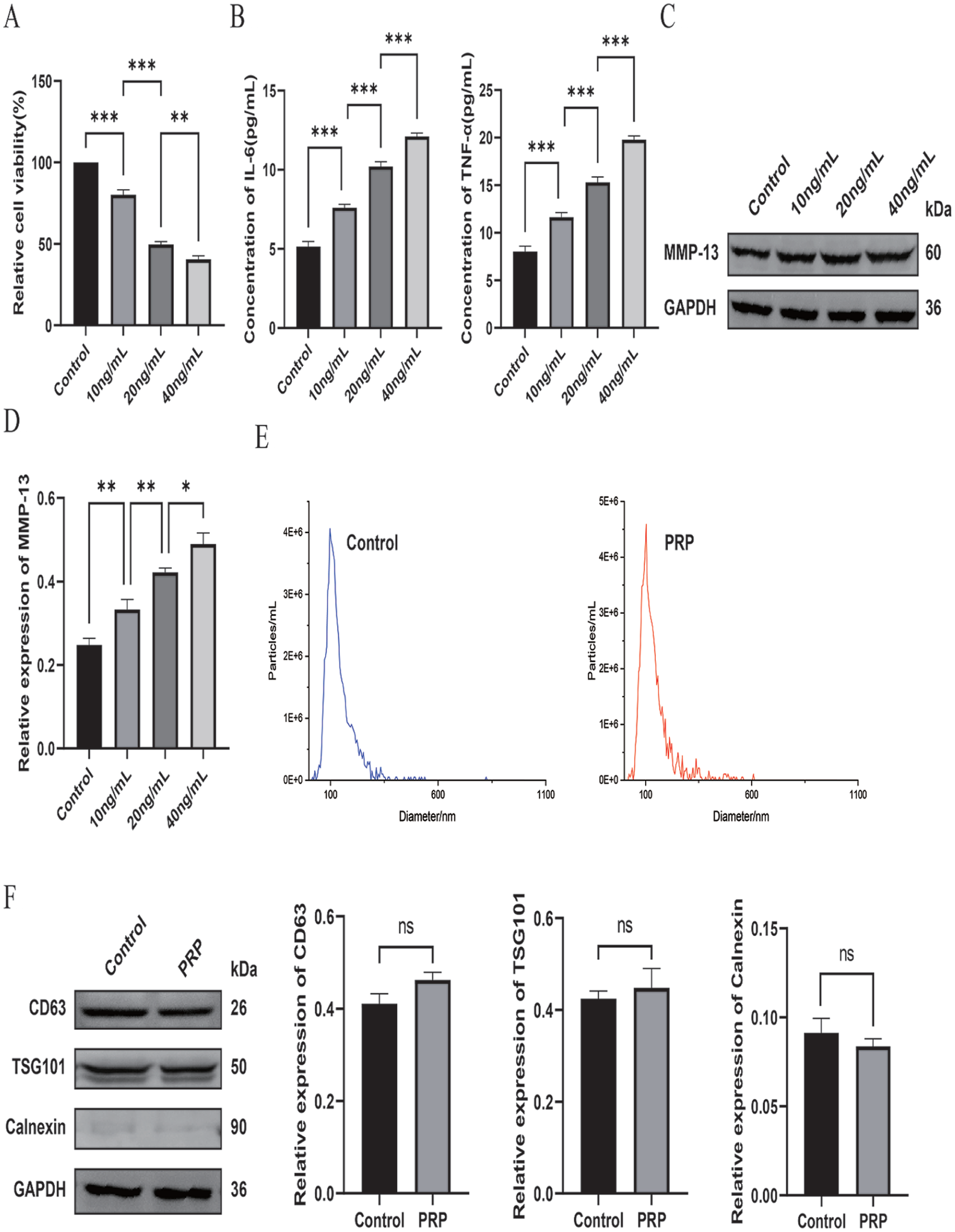
Generation of an in vitro IL-1β–triggered inflammatory damage model using C28/I2 chondrocytes and characterization of exosomes isolated from PRP-treated ADSCs.
Exosomes were subsequently isolated from PRP-treated ADSCs using differential ultracentrifugation. NTA showed that the obtained vesicles displayed a typical exosomal size distribution, predominantly ranging from 100 to 150 nm
PRP-Derived ADSC Exosomes Attenuate IL-1β–Induced Chondrocyte Injury by Upregulating LINC01106
We first investigated whether PRP exposure modulates LINC01106 expression in ADSCs. Quantitative real-time polymerase chain reaction (qRT-PCR) analysis revealed a robust elevation of LINC01106 levels following PRP treatment
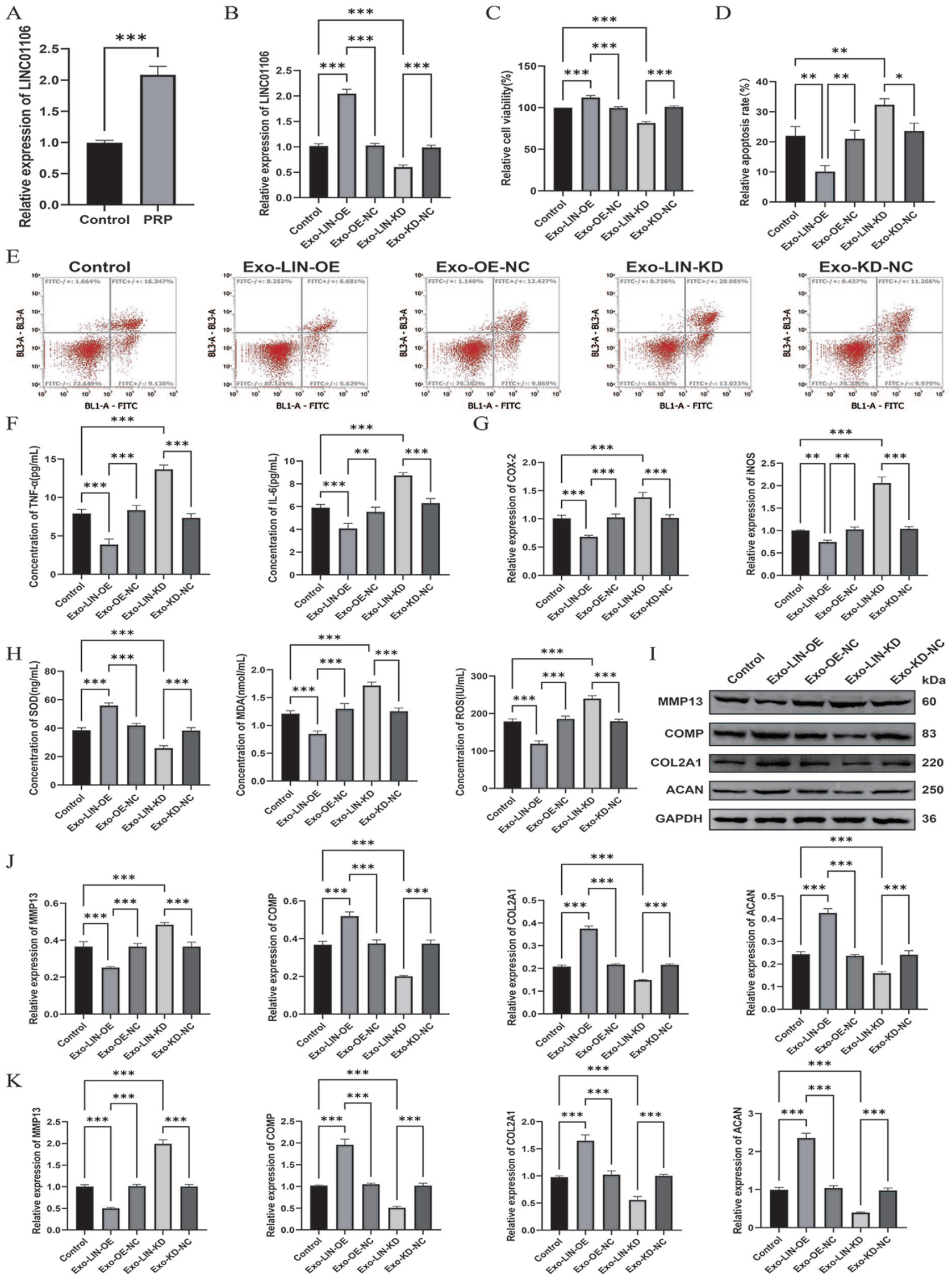
Exosomes from PRP-treated ADSCs alleviate IL-1β-induced chondrocyte injury via LINC01106.
Functional assays demonstrated that co-incubation with Exo-LIN-OE significantly rescued IL-1β-induced cytotoxicity, as reflected by enhanced cell viability and a reduced rate of apoptosis, whereas Exo-LIN-KD intensified chondrocyte injury
Matrix metabolism analyses revealed that Exo-LIN-OE effectively inhibited IL-1β-driven upregulation of MMP-13 and promoted the synthesis of major cartilage matrix proteins, including COMP, COL2A1, and ACAN. Conversely, Exo-LIN-KD accelerated matrix catabolism
LINC01106 Regulates the miR-34a-5p/SIRT1 Axis to Suppress Activation of NF-κB and MAPK Signaling Pathways
To elucidate the molecular mechanism underlying the protective role of LINC01106, downstream pathways were investigated. Based on the ceRNA hypothesis, we assessed miR-34a-5p and its validated target SIRT1. Both qRT-PCR and immunoblotting results demonstrated that Exo-LIN-OE markedly reduced miR-34a-5p expression while simultaneously restoring SIRT1 transcription and protein abundance in C28/I2 chondrocytes exposed to IL-1β. By comparison, Exo-LIN-KD triggered the opposite expression pattern
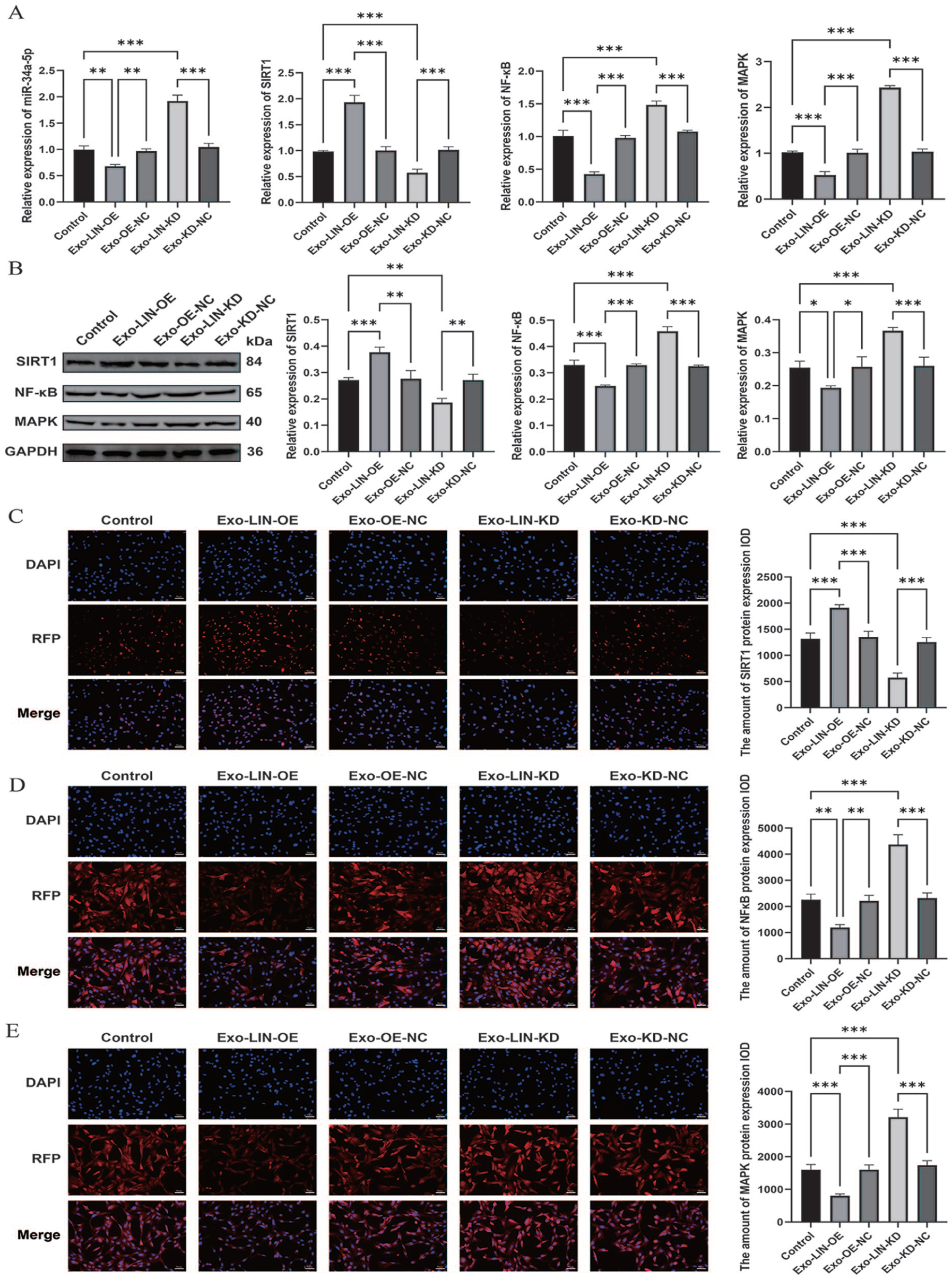
miR-34a-5p/SIRT1–dependent control of NF-κB and MAPK pathways mediated by LINC01106.
Given that SIRT1 acts as an established suppressor of NF-κB and MAPK signaling cascades, we further evaluated these pathways. Exo-LIN-OE significantly repressed IL-1β-triggered activation of both NF-κB and MAPK, whereas LINC01106 depletion enhanced pathway activation
Taken together, these results demonstrate that LINC01106-enriched exosomes from PRP-treated ADSCs mitigate IL-1β–driven chondrocyte dysfunction by functioning as a competitive sponge for miR-34a-5p, restoring SIRT1 expression, and thereby suppressing NF-κB and MAPK inflammatory signaling, ultimately contributing to the preservation of cartilage homeostasis and delaying OA progression.
LINC01106 Participates in Post-Transcriptional Regulation of miR-34a-5p
To validate whether LINC01106 serves as a ceRNA regulating miR-34a-5p, a dual-luciferase reporter system was conducted. Introduction of the miR-34a-5p mimic significantly diminished luciferase signals derived from the LINC01106 wild-type construct (LIN-WT), while the mutant construct (LIN-MUT) exhibited no detectable response
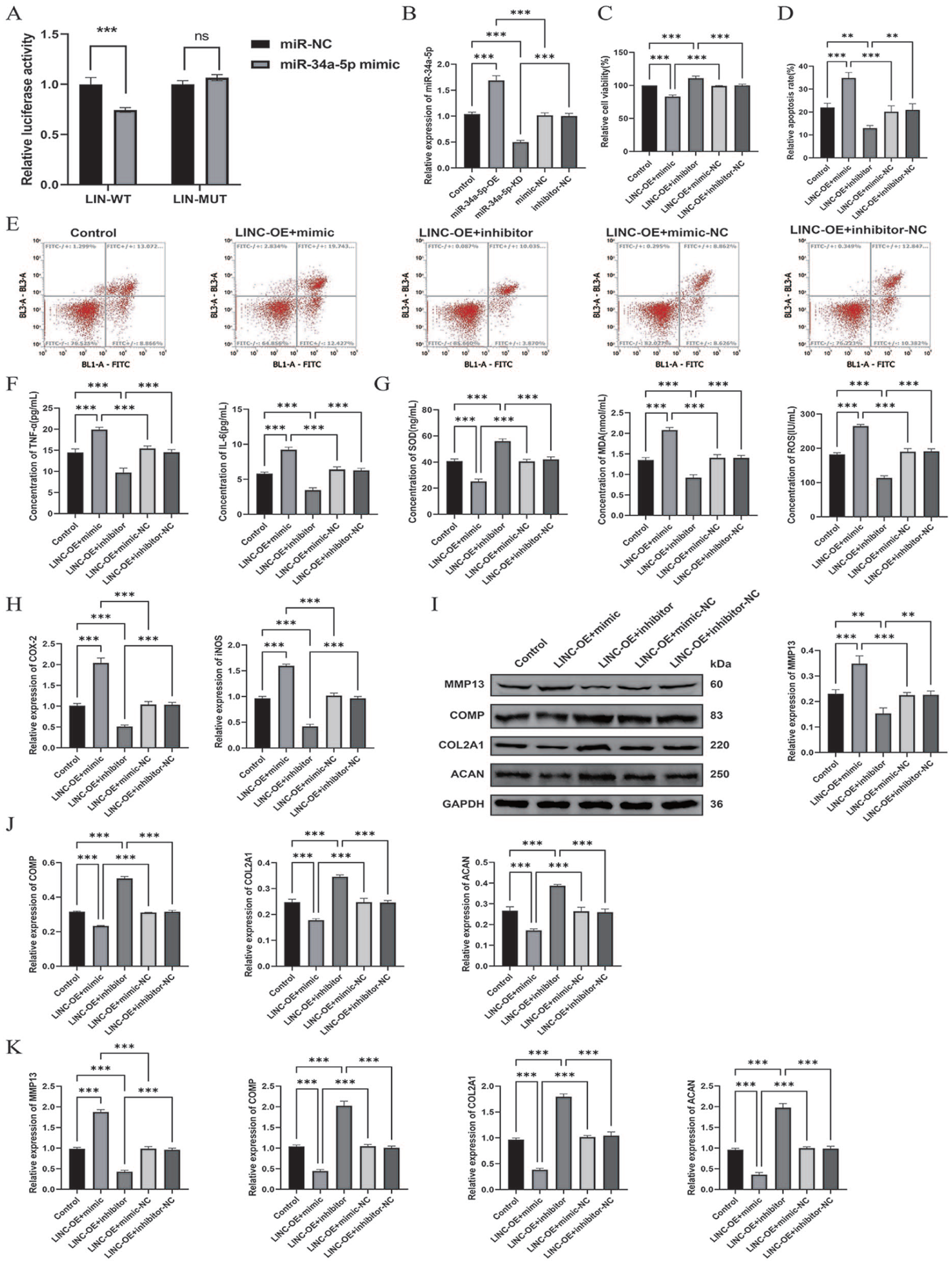
LINC01106 exerts its protective function by sequestering miR-34a-5p.
To determine whether the biological effects of LINC01106 depend on miR-34a-5p, rescue experiments were carried out in C28/I2 cells. Transfection with miR-34a-5p mimic or inhibitor effectively altered intracellular miRNA levels
LINC01106 Modulates Inflammatory Signaling Via the miR-34a-5p/SIRT1 Axis
To dissect the molecular events downstream of the LINC01106/miR-34a-5p axis, SIRT1 expression and inflammatory pathway activation were evaluated. C28/I2 chondrocytes were initially transfected with either a miR-34a-5p mimic or inhibitor and subsequently treated with Exo-LIN-OE. Molecular analyses revealed that enforced miR-34a-5p expression negated Exo-LIN-OE–driven SIRT1 elevation and restored NF-κB and MAPK signaling to an IL-1β–like activation state
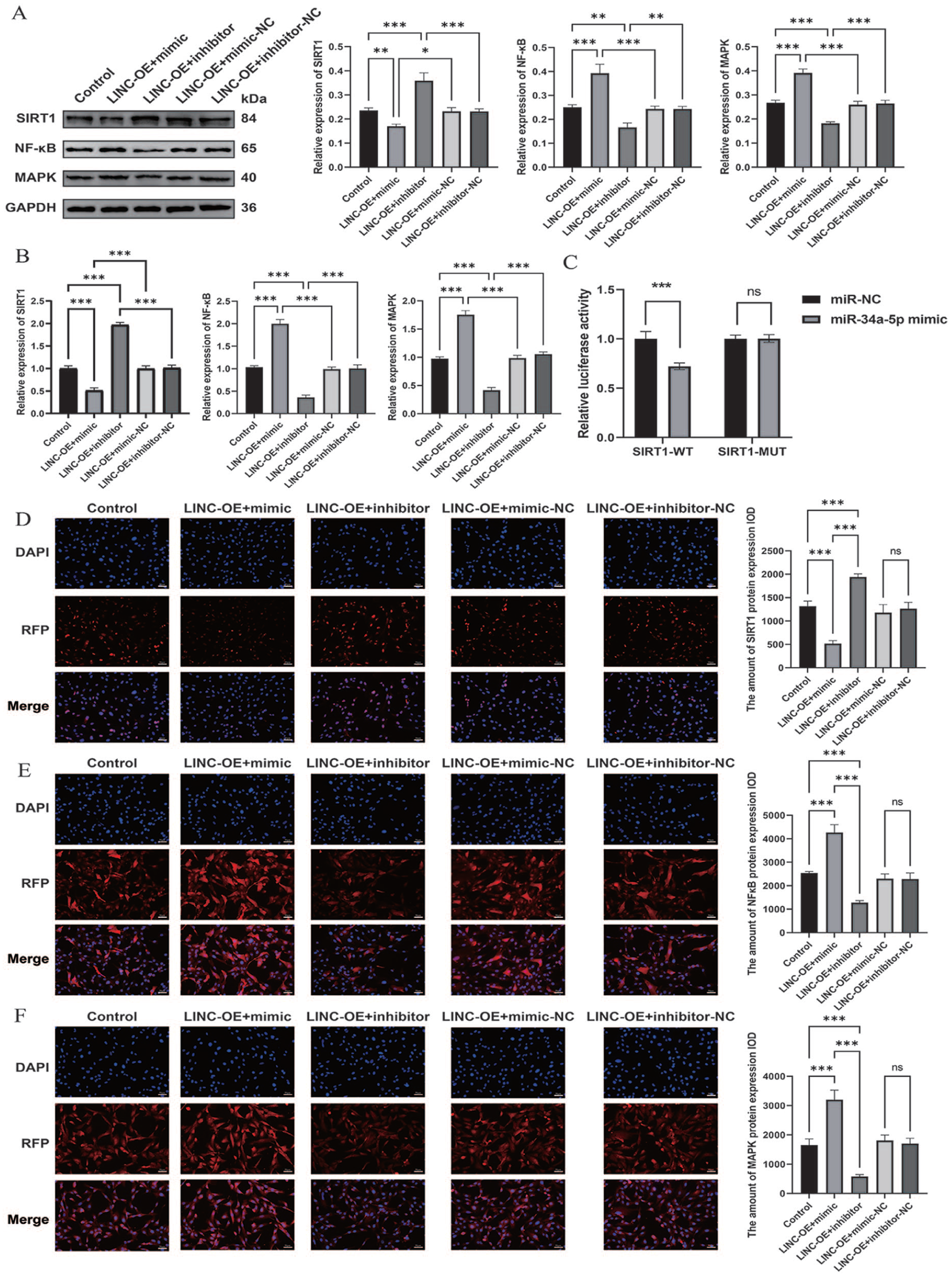
LINC01106 inhibits inflammatory signaling pathways by targeting SIRT1 through miR-34a-5p.
To verify whether SIRT1 is directly regulated by miR-34a-5p, a dual-luciferase reporter assay was conducted. Forced expression of miR-34a-5p markedly reduced luciferase activity driven by the SIRT1-WT, whereas the MUT construct showed no detectable response
Collectively, these results establish the miR-34a-5p/SIRT1 axis as a key mediator of Exo-LIN-OE–induced anti-inflammatory effects through coordinated suppression of NF-κB and MAPK cascades.
Discussion
Our findings identify, for the first time, a PRP-ADSC exosome–based mechanism in which LINC01106 attenuates chondrocyte injury in OA by fine-tuning the miR-34a-5p/SIRT1 axis. Our data demonstrate that PRP stimulation markedly increases LINC01106 abundance in ADSC-derived exosomes, and that these LINC01106-rich vesicles exert potent protective effects against IL-1β–induced inflammatory damage. Specifically, exosomal LINC01106 attenuated apoptosis, inflammation, oxidative stress, and extracellular matrix degradation, while simultaneously enhancing chondrocyte proliferation. Collectively, these findings highlight LINC01106-containing exosomes as promising cell-free therapeutic vectors for OA.
Growing evidence supports the pivotal role of lncRNAs as prognostic biomarkers and regulatory molecules across diverse pathologies.32 -34 Recent studies have identified LINC01106 as a novel lncRNA aberrantly expressed in multiple cancers. Guo et al. 35 demonstrated that LINC01106 enhances colorectal cancer cell proliferation and stem-like properties by engaging a Gli-dependent positive feedback mechanism. Similarly, Hong et al. 18 found that silencing LINC01106suppresses gastric cancer cell growth, migratory capacity, and facilitates invasion through miR-34a-5p/MYCN–dependent signaling. In addition, LINC01106 has been shown to promote tumor angiogenesis by influencing the miR-449b-5p/VEGFA pathway. 36 However, its relevance in OA has not been previously explored. Our findings show that PRP significantly upregulates LINC01106 in ADSCs and that exosomes enriched in LINC01106 exert potent chondroprotective effects, suggesting that LINC01106 may serve as an effective molecular cargo in exosome-based regenerative therapy.
To further elucidate its biological role, we examined the effects of modulating LINC01106 expression in ADSC-derived exosomes. Consistent with the clinical observation that IL-1β promotes catabolic chondrocyte responses, our in vitro results show that LINC01106 overexpression suppresses apoptosis, reduces inflammatory cytokine secretion, ameliorates oxidative stress, and protects against extracellular matrix degradation. These data reinforce the concept that LINC01106 functions as a protective regulator during OA progression. Accumulating studies indicate that lncRNAs often act as ceRNAs to sequester miRNAs through shared miRNA response elements.16,17,37 The lncRNA-miRNA-mRNA regulatory network is increasingly regarded as a pivotal mechanism underlying the pathogenesis of numerous disorders, including OA.38 -40 In our study, bioinformatic prediction and luciferase assays confirmed miR-34a-5p interacts with LINC01106. Moreover, LINC01106 was found to suppress miR-34a-5p expression in chondrocytes, thereby relieving its repression of SIRT1. Consequently, LINC01106 enhanced SIRT1 levels and attenuated apoptosis, inflammation, and matrix degradation. Notably, the protective actions of LINC01106-enriched exosomes were abrogated by miR-34a-5p re-expression or SIRT1 knockdown. Taken as a whole, these observations suggest that the chondroprotective role of LINC01106 is largely dependent on its ability to neutralize miR-34a-5p.
Accumulating evidence underscores miR-34a-5p as a pivotal regulator in OA progression. According to Endisha et al., 23 miR-34a-5p is markedly upregulated in synovial fluid of patients with advanced-stage OA, where it accelerates joint degeneration by inducing chondrocyte apoptosis and facilitating extracellular matrix degradation. Another study showed that miR-34a-5p contributes to cartilage degeneration by promoting chondrocyte apoptosis and senescence via DLL1 suppression and PI3K/AKT signaling cascade modulation. 41 Furthermore, impairment of the miR-34a-5p/SIRT1/p53 signaling axis under oxidative stress conditions has been implicated in sustained chondrocyte injury and inflammatory activation. 42 Recent evidence also demonstrated that downregulation of the lncRNA SNHG7 promotes OA progression, as SNHG7 confers chondroprotection through miR-34a-5p sequestration and regulation of SYVN1 expression. 43 In agreement with these reports, our results show that restoring miR-34a-5p or silencing SIRT1 reverses the beneficial effects mediated by LINC01106-rich exosomes, further confirming the pathogenic significance of miR-34a-5p in OA.
We then examined the signaling molecules acting downstream of miR-34a-5p, confirming SIRT1 as a major effector in this regulatory pathway. SIRT1, an NAD⁺-dependent deacetylase, is known to regulate numerous cellular processes and is critically involved in preserving tissue homeostasis.21,22 A substantial body of evidence supports the protective function of SIRT1 in preserving cartilage integrity and alleviating OA progression.44,45 Matsuzaki et al. 46 demonstrated that chondrocyte-specific deletion of SIRT1 accelerates OA progression in mice under mechanical stress and aging. Moon et al. 47 further reported that SIRT1 deacetylates and inactivates NF-κB p65, thereby exerting anti-inflammatory effects in chondrocytes. Independent studies have confirmed that SIRT1 attenuates pro-inflammatory and catabolic pathways, including NF-κB and MAPK, during OA development.48,49 These observations, together with our findings, support the conclusion that the LINC01106/miR-34a-5p/SIRT1 regulatory axis is a major determinant of chondrocyte fate under inflammatory conditions.
Another key discovery of our study is that PRP enhances the bioactivity of ADSC-derived exosomes. PRP is increasingly recognized as a therapeutic option for OA, and PRP-derived exosomes have demonstrated superior regenerative efficacy compared with PRP alone.11,12,14 According to Zhang et al., 50 PRP-originated exosomes facilitate chondrocyte growth and reduce apoptosis by modulating Wnt/β-catenin signaling cascade. Additional reports indicate that PRP enhances exosome secretion and functional potency in stem cells.51,52 Our results extend these observations by showing that PRP pretreatment selectively enriches LINC01106 in ADSC-derived exosomes, thereby augmenting their ability to regulate the miR-34a-5p/SIRT1 axis and protect chondrocytes. This mechanistic understanding offers a solid theoretical foundation for the improved therapeutic performance of PRP-ADSC combination strategies.
We also identified NF-κB and MAPK pathways as downstream targets of the LINC01106/miR-34a-5p/SIRT1 cascade. These inflammatory cascades are well-established contributors to OA pathogenesis by promoting cytokine expression and matrix degradation.53,54 SIRT1-mediated deacetylation of NF-κB p65 and its regulatory effects on MAPK phosphorylation represent crucial anti-inflammatory mechanisms in chondrocytes. Our immunofluorescence and Western blot results confirm that LINC01106-rich exosomes markedly suppress NF-κB and MAPK activation in IL-1β-treated chondrocytes by restoring SIRT1 levels via miR-34a-5p inhibition. These findings collectively support the feasibility of therapeutically exploiting the LINC01106/miR-34a-5p/SIRT1 axis in OA. It should be noted that the present mechanistic analysis focused primarily on NF-κB and MAPK signaling pathways, which are well-established mediators of OA pathophysiology. However, additional pathways, including PI3K/AKT signaling and autophagy-related mechanisms, may also contribute to the observed chondroprotective effects. For instance, SIRT1 has been reported to directly activate autophagy in chondrocytes, 44 and the PI3K/AKT pathway has been implicated in miR-34a-5p–mediated chondrocyte apoptosis. 41 Future studies should explore whether the LINC01106/miR-34a-5p/SIRT1 axis also modulates these additional pathways to provide a more comprehensive mechanistic picture.
Nevertheless, several limitations of the present study warrant consideration. First, all experiments were conducted in vitro using IL-1β–stimulated C28/I2 chondrocytes, and the absence of in vivo validation in established OA animal models (e.g., destabilization of the medial meniscus or collagenase-induced models) limits the extrapolation of the chondroprotective findings to a therapeutic context. Future studies employing intra-articular delivery of LINC01106-enriched exosomes in animal models are warranted to assess efficacy, biodistribution, and safety. Second, LINC01106 may regulate additional miRNAs or pathways, and miR-34a-5p may act on other targets beyond SIRT1. A more comprehensive analysis is needed to elucidate the entire regulatory network. Third, the exclusive use of the immortalized C28/I2 cell line, maintained in monolayer culture, may not fully recapitulate the phenotypic characteristics of primary human OA chondrocytes; future work should validate key findings in patient-derived primary chondrocytes or 3-dimensional culture systems. Fourth, PRP preparations were not systematically characterized for platelet concentration or growth factor content across batches, which may introduce inter-experimental variability in exosomal yield and cargo composition. In addition, direct evidence of exosome internalization was not provided; uptake was inferred from functional outcomes, and future studies should employ fluorescent labeling combined with confocal microscopy for direct visualization. Last, the upstream signaling mechanisms by which PRP upregulates LINC01106 in ADSCs remain unclear and warrant further investigation.
In summary, our study identifies LINC01106 as a previously unrecognized chondroprotective lncRNA enriched in PRP-primed ADSC exosomes. We demonstrate that LINC01106 protects chondrocytes from IL-1β–triggered injury via repression of miR-34a-5p, restoring SIRT1, and inhibiting NF-κB and MAPK signaling cascades. Furthermore, targeting miR-34a-5p or enhancing SIRT1 reverses the detrimental effects associated with LINC01106 deficiency. These results offer novel mechanistic perspectives on the pathogenesis of OA and position LINC01106-rich exosomes as a promising cell-free therapeutic strategy deserving further preclinical and clinical evaluation.
Footnotes
Acknowledgements
The authors have no acknowledgments to report.
Ethical Considerations
Not applicable.
Informed Consent
Not applicable.
Consent for Publication
Not applicable.
Author Contributions
Xuan Zhang is responsible for the guarantor of integrity of the entire study, study concepts & design, literature research, data acquisition & analysis, manuscript preparation & editing & review; Wentao Liu is responsible for the guarantor of integrity of the entire study, definition of intellectual content, clinical studies, experimental studies, data analysis, statistical analysis; Jinke Ren is responsible for the data analysis, manuscript preparation & editing; Yangyi Yu is responsible for the definition of intellectual content, literature research, clinical studies, manuscript review; Zhongshi Xu is responsible for the guarantor of integrity of the entire study, study concepts, definition of intellectual content. All authors read and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study is funded by The Social Development Science and Technology Plan Project of Heyuan City, Acceptance Number: 241031161472607, Project Number: Hebei Science and Technology Social Development 122.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets used or analyzed during the current study are available from the responding author on reasonable request.
Clinical Trial Number
Not applicable.
Clinical Trial Registration
Not applicable.