
Abstract
Second malignant neoplasms (SMNs) are rare but serious late effects in childhood acute lymphoblastic leukemia (ALL) survivors. Mature peripheral T-cell lymphomas involving both the breast and ovary are exceedingly rare, and their relationship with germline predisposition variants remains poorly understood. We report a 17-year-old female, previously treated for high-risk B-cell ALL and in sustained remission, who developed bilateral breast and ovarian masses in 2024. Histopathology and immunohistochemistry confirmed a mature T-cell lymphoma (CD7+, CD99+, weak CD5/CD3, Ki-67 >90%, TdT–). Whole-exome sequencing revealed a germline pathogenic BRCA2 variant (c.1910-2A>T) along with additional likely pathogenic variants (CUX1, MED12L, POLR3B, PIK3CA). The disease progressed rapidly despite CHOP chemotherapy, and the patient died shortly after diagnosis. This report describes an exceptionally rare SMN of peripheral T-cell lymphoma with breast and ovarian involvement in a pediatric leukemia survivor. The coexistence of a germline BRCA2 mutation and additional genomic alterations suggests a multigenic predisposition hypothesis. Early incorporation of next-generation sequencing may uncover molecular vulnerabilities and inform alternative therapeutic strategies in refractory hematologic malignancies.
Keywords
Introduction
Although rare, relapse and/or development of SMNs in acute lymphoblastic leukemia (ALL) survivors represent a significant diagnostic and therapeutic challenge. The cumulative risk for SMN within the first 20 years after diagnosis remains low but is clinically relevant and influenced by treatment intensity and genetic susceptibility.1,2
SMN patients may exhibit persistent chromosomal instability, including non-clonal chromosomal aberrations, whose frequency and type depend on prior therapy and hematopoietic lineage. 3 Additional late effects include cardiac toxicity, neurotoxicity, hepatic or bone damage, infertility, and neurocognitive decline.4–6 In this context, T-cell lymphomas in extranodal sites such as breast and ovary are exceedingly rare, and their occurrence in patients with germline BRCA2 variants remains poorly understood. 7 Germline pathogenic variants in BRCA2 are classically associated with epithelial tumors, but recent evidence suggests a role in hematopoietic dysfunction. 8 Here, we describe a rare SMN of peripheral T-cell lymphoma with synchronous breast and ovarian involvement in a survivor of pediatric leukemia harboring a germline BRCA2 variant.
Case presentation
A 17-year-old female with a history of high-risk B-cell acute lymphoblastic leukemia (B-ALL) diagnosed at age 12 was treated with multi-agent chemotherapy according to the Total XIII-B protocol, a St. Jude–based risk-adapted pediatric ALL regimen, including multi-agent chemotherapy with high-dose methotrexate and cytarabine during consolidation. She achieved complete remission with sustained negative minimal residual disease (MRD) documented in March 2023.
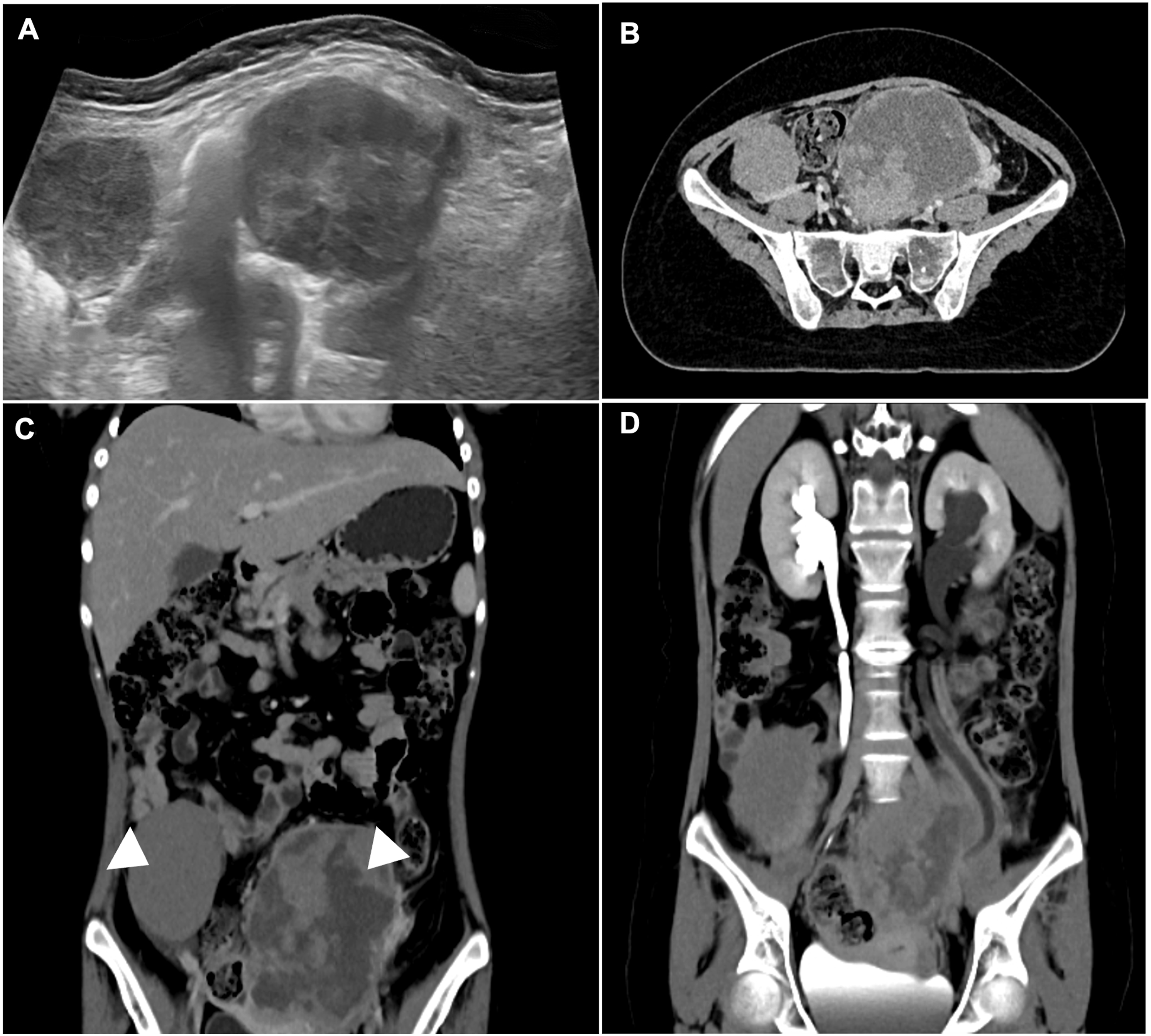
Approximately three months after remission was documented, the patient developed progressive bilateral breast and adnexal masses. Physical examination revealed firm, immobile nodules in both breasts and large pelvic tumors. Breast and pelvic ultrasound (Figure 1(a) and (b)) and thoracic CT (Figure 1(c) and (d)) confirmed bilateral breast involvement. Abdominal ultrasound and CT demonstrated large bilateral ovarian masses with associated lymphadenopathy and renal compromise (Figure 2(a)–(d)). Laboratory tests showed markedly elevated lactate dehydrogenase (LDH) (1649 U/L) and normal uric acid. Serum tumor markers (AFP, β-hCG, CEA, CA-15-3, CA-125, CA 19-9) were within reference ranges. Bilateral breast lymphoma presenting as SMNs. Bilateral ovarian lymphoma with lymphatic dissemination and urological compromise due to mass effect.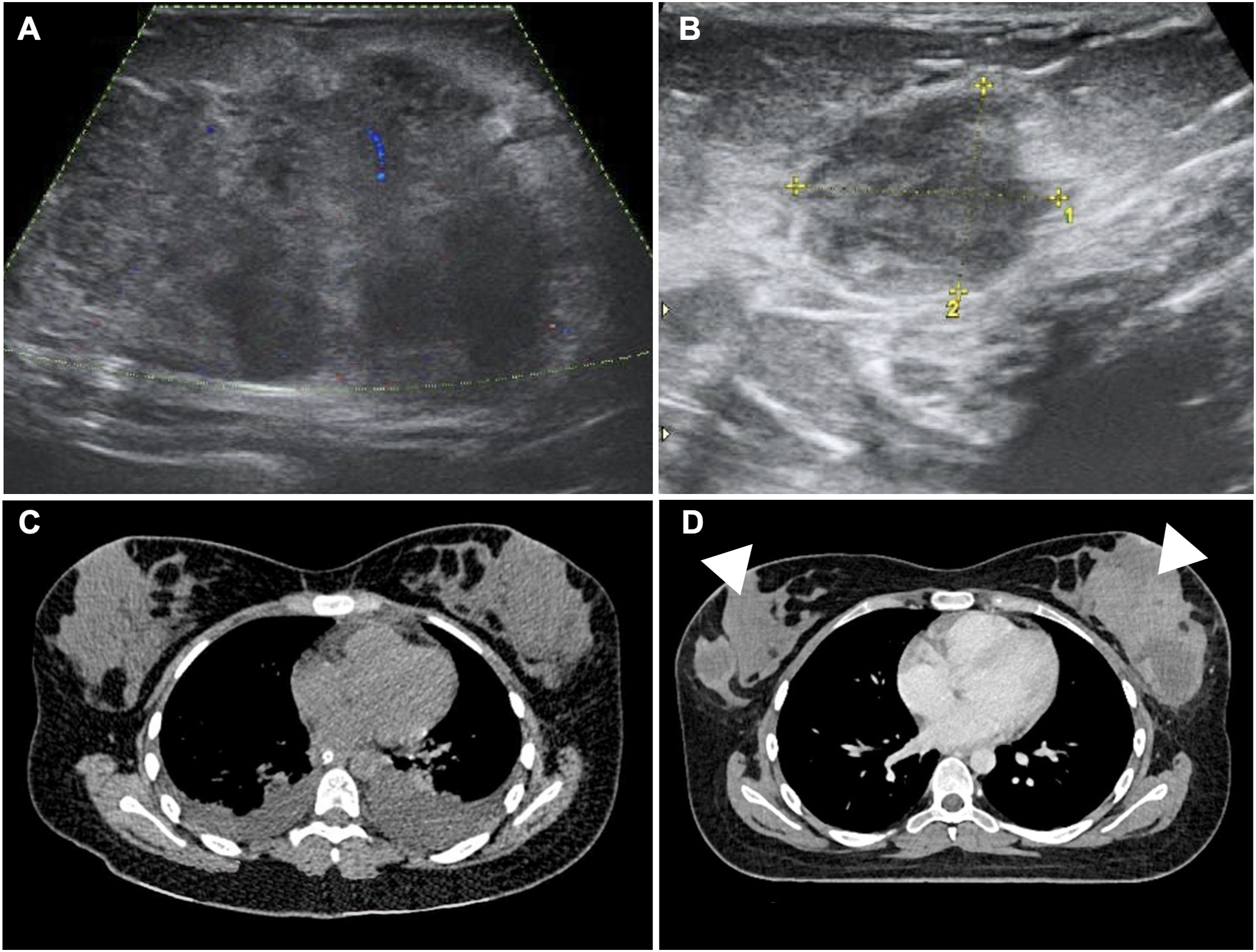
Histopathological examination of breast biopsies demonstrated malignant lymphoid infiltration. Immunohistochemistry showed CD7 and CD99 positivity, weak CD5/CD3 expression, and negativity for TdT, PAX5, CD10, and CD20, with Ki-67 >90%. This immunophenotypic profile was compatible with a mature T-cell lymphoma, most consistent with peripheral T-cell lymphoma, not otherwise specified. In the context of disseminated disease with synchronous ovarian masses, surgical resection of the ovarian mass was subsequently performed. Given the disseminated disease with synchronous ovarian masses and the associated obstructive uropathy with loss of left renal function, surgical resection of the ovarian mass was subsequently performed for diagnostic and therapeutic purposes. Histopathological examination of the ovarian specimen demonstrated an ovoid mass measuring 10 cm in greatest dimension, with a whitish-gray, dull external surface and medium consistency with congestive areas (Figure 3(a) and (b)). Microscopically, there was diffuse effacement of the normal ovarian architecture by a monotonous lymphoid proliferation replacing the entire ovarian parenchyma. At higher magnification, the tumor was composed of atypical medium-to-large lymphoid cells with irregular nuclei, a high nuclear-to-cytoplasmic ratio, and occasional prominent nucleoli, with abundant mitotic and apoptotic figures (Figure 3(c) and (d)), consistent with an aggressive lymphoid neoplasm. Ovarian involvement by aggressive lymphoid neoplasm.
Germline whole-exome sequencing (WES) of peripheral blood revealed a pathogenic BRCA2 splice-site variant (c.1910-2A>T) together with additional likely pathogenic variants in CUX1, MED12L, POLR3B, and PIK3CA, suggesting multigenic predisposition.
The patient received one cycle of CHOP (Cyclophosphamide + Hydroxydaunorubicin + Oncovin + Prednisone) chemotherapy but exhibited primary refractory disease. Rapid progression with pancytopenia and infectious complications ensued, and she died within weeks of diagnosis despite supportive care.
Discussion
This case illustrates an exceptionally rare second malignant neoplasm in a pediatric ALL survivor, presenting as peripheral T-cell lymphoma with synchronous involvement of the breast and ovary. While breast lymphomas are already uncommon, fewer than 5% are of T-cell lineage, and ovarian involvement is even rarer due to the paucity of lymphoid tissue in that organ.9–11 The bilateral pattern of presentation without clinically evident peripheral nodal disease, as shown in the imaging studies, emphasizes the aggressive extranodal nature of this tumor.
Immunophenotypic characterization was essential to establish the diagnosis. The neoplastic cells expressed CD7 and CD99 with weak CD5/CD3, while negative for TdT, PAX5, CD10, and CD20, with a high Ki-67 proliferation index (>90%). This immunoprofile supported a mature T-cell lineage; however, complete exclusion of precursor T-cell neoplasms was limited by the absence of additional studies. 12 The absence of TdT expression is consistent with a mature T-cell neoplasm,13–17 and may reflect therapy-induced clonal evolution. This finding, together with the patient’s therapeutic history, indicates that a lineage switch or clonal divergence could not be confirmed, as no comparative molecular analysis between the initial B-ALL and the subsequent lymphoma was available under treatment pressure. 18
From a clinical perspective, synchronous breast and ovarian involvement creates a complex differential diagnosis. Previous reports, such as those by Chishima et al. describing bilateral breast and ovarian Burkitt lymphoma, illustrate that aggressive lymphomas can present in this pattern. 9 However, in contrast to those B-cell cases, our patient exhibited a T-cell lineage, underscoring the exceptional nature of this presentation.
Therapeutically, the primary refractory response to CHOP chemotherapy highlights the poor prognosis of peripheral T-cell lymphomas, especially in adolescents.14,16 Alternative regimens such as CHOEP (CHOP + Etoposide), targeted therapy (e.g., brentuximab in CD30+ cases), or enrollment in clinical trials may be required in similar scenarios, although access is often limited. In this patient, rapid progression precluded the application of additional lines of treatment. 14
The genomic context provides an additional layer of complexity. Whole-exome sequencing revealed a pathogenic germline BRCA2 splice-site variant, along with other likely pathogenic variants (CUX1, MED12L, POLR3B, PIK3CA). The history of hyperleukocytosis at ALL diagnosis, the exposure to intensive therapy, and the presence of germline genomic alterations together create a background of high genomic instability that may favor the emergence of atypical secondary hematologic malignancies.8,19–21 CUX1 is a recognized haploinsufficient tumor suppressor gene frequently inactivated in acute myeloid leukemia and therapy-related myeloid neoplasms, where its loss disrupts DNA repair and promotes epigenetically driven genomic instability.22,23 Alterations in the PI3K/AKT/mTOR signaling pathway, often involving PIK3CA, play a central role in lymphoid cell survival, proliferation, and therapy resistance and represent established therapeutic targets in lymphoma.24–26 MED12L belongs to the Mediator complex family, a key regulator of transcription and chromatin organization, whose dysregulation has been implicated in leukemic chromatin remodeling and transcriptional instability.27,28 Finally, POLR3B encodes a subunit of RNA polymerase III, and mutations affecting RNA polymerase subunits have been shown to disrupt transcriptional homeostasis and cellular growth control in genetic diseases and cancer.29,30 Collectively, these alterations are unlikely to represent direct oncogenic drivers, but may establish a permissive genomic background that predisposes hematopoietic cells to genomic stress, clonal evolution, and malignant transformation under genotoxic pressure.
In this context, although causal inference is not possible in a single-patient report, a plausible mechanistic explanation for the development of a secondary lymphoid neoplasm involves the convergence of therapy-related genomic stress and inherited DNA-repair vulnerability. Intensive cytotoxic exposure during ALL treatment can promote persistent genomic instability, including the accumulation of nonclonal chromosomal aberrations and broader ‘genome chaos’, creating an evolutionary substrate from which aggressive secondary clones may emerge through selection and adaptation. 3 In parallel, germline BRCA2 loss-of-function compromises homologous recombination–mediated repair of DNA double-strand breaks, increasing reliance on error-prone repair pathways and favoring chromosomal rearrangements under genotoxic pressure.7,20 Together, these processes provide a coherent model in which therapy-induced genomic instability is amplified by impaired BRCA2-mediated DNA repair, facilitating clonal diversification and malignant transformation. Consistent with this concept, population-based data support an increased risk of hematologic malignancies among carriers of germline BRCA2 loss-of-function variants. 8
While BRCA2 is classically associated with breast and ovarian carcinoma, recent evidence supports its involvement in hematopoietic malignancies, 8 and this case may add to the growing body of evidence linking inherited predisposition to rare lymphoid neoplasms.
This case report is limited by the restricted availability of tumor material for additional ancillary studies. Following confirmatory diagnostic evaluation and subsequent transfer of the specimens to another institution for continuation of oncologic care, the tissue could not be retrieved for further analysis. Therefore, extended immunophenotypic and molecular characterization could not be performed.
Conclusions
This exceptionally rare case of peripheral T-cell lymphoma with synchronous involvement of the breast and ovary as a SMN in a pediatric ALL survivor highlights the interplay between prior intensive therapy, hyperleukocytosis at diagnosis, and inherited predisposition. The coexistence of a germline BRCA2 mutation and additional genomic alterations supports the hypothesis of multigenic predisposition, clonal evolution, and phenotypic plasticity in lymphoid transformation. The primary refractory course to CHOP, even in apparently localized disease, underscores the urgent need for precision-oncology strategies and innovative therapies. Early incorporation of next-generation sequencing and a multidisciplinary evaluation are critical to refine diagnosis, identify molecular vulnerabilities, and guide innovative management in such rare presentations.
Footnotes
Acknowledgements
The authors thank the Pathology and Oncology Departments of the Instituto Estatal de Cancerología for their collaboration in diagnostic support and patient care. We also acknowledge the Department of Hematopathology of the Instituto Nacional de Cancerología for their expert diagnostic confirmation and immunophenotypic evaluation.
Ethical considerations
This case report was reviewed according to institutional guidelines and was exempt from formal approval by the Research Ethics Committee. Written informed consent for publication was obtained from the patient’s parents.
Funding
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
All data generated or analysed during this case report are included in this published article.