
Abstract
Novel copolymers based on acrylate epoxidized soybean oil (
Introduction
Acrylate epoxidized soybean oil (
However, the polymer synthesized from AESO (
Despite their effectiveness, conventional monomers like styrene, divinylbenzene, methyl methacrylate, and isocyanatoethyl methacrylate are classified as hazardous air pollutants due to their volatility during curing and processing. Although they improve resin fluidity and facilitate composite fabrication, their environmental and health risks have prompted the search for safer alternatives. In this context, recent research has focused on developing green comonomers derived from renewable resources,7–9 such as methacrylated4,5 fatty acids and maleinated castor oil monoglycerides.
10
Moreover, the position and geometry of double bonds in these bio-based monomers—whether terminal or internal—play a crucial role in determining polymerization kinetics, crosslinking efficiency, and the final properties of the cured materials. Terminal double bonds, like those in
Among the various bio-based alternatives explored in recent years, vanillin has emerged as a particularly promising platform molecule. Derived from lignin, vanillin is one of the few commercially available mono-aromatic compounds obtained from renewable biomass at industrial scale.14–16 Its molecular structure includes both aldehyde and phenolic hydroxyl functional groups, which offer versatile sites for chemical modification, enabling the synthesis of a wide range of functional monomers. These vanillin-based monomers can be tailored to include vinyl, epoxy, methacrylate, or other reactive groups, making them suitable for incorporation into thermosetting and thermoplastic polymer networks.17–21
Importantly, the aromatic backbone of vanillin imparts rigidity and thermal stability to the resulting polymers,
21
addressing one of the key limitations of vegetable oil-based resins like
Building upon these insights, this study presents the synthesis of two bio-based vinylic aromatic isomers derived from vanillin, designed to leverage both the structural rigidity of the aromatic backbone and the reactivity of vinyl functionalities. The synthesis of vanillin-derived vinylic aromatic monomers through the functionalization of vanillyl alcohol with 4-vinylbenzyl chloride (terminal double bond) and cinnamyl bromide (internal double bond) introduces a distinctive structural framework. Each resulting monomer contains three aromatic rings in total: the original vanillin ring plus the additional aromatic moieties from the vinylbenzyl or cinnamyl substituents. The resulting monomers represent a structurally reinforced class of bio-based copolymers. Their triple aromatic backbone and tunable vinyl functionality provide a pathway to materials with higher glass transition temperatures, and enhanced degradation resistance compared to conventional
Experiment
Materials
Vanillin, cinnamyl bromide (purity ≥97%), 4-vinylbenzyl chloride (purity ≥90%), Acrylate-Epoxidized Soybean Oil (
Characterization
1H-NMR spectra were recorded using a Bruker spectrometer operating at 300 MHz. Deuterated dimethyl sulfoxide (DMSO-d6) was used as the solvent for vanillin derivatives, while deuterated chloroform (CDCl3) was employed for
FTIR-HATR spectra were obtained using an IRPrestige-21 SHIMADZU spectrometer equipped with a Horizontal Attenuated Total Reflectance (HATR) accessory featuring a diamond crystal. Spectra were collected over the range of 4000–560 cm-1 with a resolution of 4 cm-1 and averaged over 64 scans.
Thermal analysis (DSC-TGA) was conducted using a PerkinElmer Simultaneous Thermal Analyzer (STA 8000) under a nitrogen purge (20 mL/min) at a heating rate of 10°C/min, covering a temperature range from 30 to 650°C.
DSC curves for the polymer samples were recorded using a TA Instruments Discovery 250 calorimeter under a nitrogen atmosphere. For glass transition temperature (Tg) determination, approximately 5 mg of each sample was first heated from 25 to 175°C at 10°C/min to erase thermal history. The samples were then cooled to −50°C at the same rate, followed by a second heating cycle from −50 to 175°C. Tg values were extracted from the second heating curve.
Swelling testing. Swelling experiments were performed by gravimetric method performed in three different solvents: THT, Toluene and DMF. Circular polymer specimens (2.5 cm in diameter, 2 mm thick) were prepared (as following recommendations from polymer testing standards, 26 weighed (initial mass, (mi), and immersed in solvent for 24 h. After immersion, the samples were gently dried to remove excess solvent and immediately re-weighed to record the final mass, (mf).
Synthesis of vanillyl alcohol from vanillin
Vanillyl alcohol was synthesized following a microscale method 27 with modifications 28 to improve the yield (68.3%) and adapt the procedure to a larger scale. Vanillin (10 g, 0.066 mol) was dissolved in 20 mL of ethanol in a beaker. Subsequently, 9.6 mL of sodium borohydride solution (3.4 M in 1.0 M NaOH) was added dropwise under magnetic stirring. The addition was carried out slowly due to the exothermic nature of the reaction.
After complete addition of the NaBH4 solution, the reaction mixture was placed in an ice bath and stirred for 20 min. It was then removed from the ice bath and stirred at room temperature for an additional 10 min to ensure completion of the reduction. The mixture was returned to the ice bath, and 6 M hydrochloric acid was added dropwise with continuous stirring until hydrogen gas evolution ceased, the pH became acidic, and a precipitate formed.
The resulting precipitate was collected by vacuum filtration, washed thoroughly with distilled water, and dried in a desiccator for at least 24 h. The crude vanillyl alcohol was then recrystallized from toluene to obtain the purified product, with a yield of 76%.
Synthesis of monomer vinylic aromatic derivatives of vanillin
The synthesis of vinylic aromatic monomers derived from vanillin (Figure 1) was carried out as follows. A two-neck round-bottom flask was charged with 0.058 mol (6.55 g) of potassium tert-butoxide, 65 mL of tetrahydrofuran (THF), and 30 mL of tert-butanol as solvents. The flask was placed in a high-frequency ultrasonic bath for 10 min to promote dissolution and activation. It was then equipped with a magnetic stirrer and transferred to an oil bath at reflux temperature. Chemical structure of vinylic aromatic derivates of vanillin.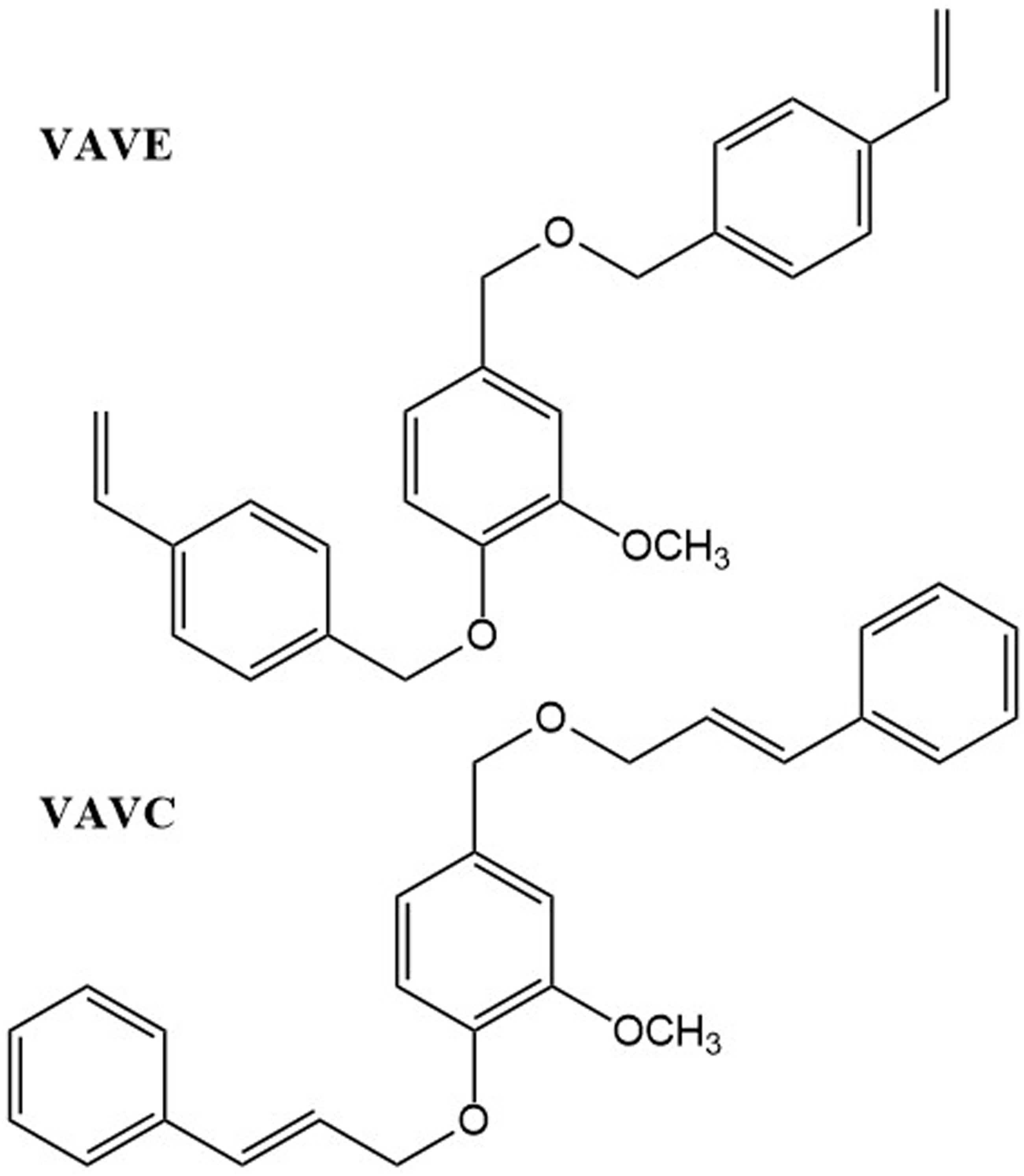
Separately, 0.019 mol (3 g) of vanillyl alcohol was dissolved in 20 mL of THF. This solution was added slowly to the potassium tert-butoxide mixture under constant stirring. A precipitate formed due to the deprotonation of vanillyl alcohol. After 20 min of stirring, 0.043 mol (7.3 g) of 4-vinylbenzyl chloride or 0.043 mol (8.7) g of cinnamyl bromide were added to produce
After completion, the reaction was cooled to 50°C and concentrated under reduced pressure. Then, 90 mL of purified water was added, and the aqueous phase was extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over magnesium sulfate heptahydrate (MgSO4·7H2O) and concentrated under vacuum.
The crude product was purified by column chromatography using silica gel and a toluene/hexane mixture (70:30 v/v) as the eluent.
Synthesis of polymers
Composition of polymer mixtures and DSC data: initial (Ti), final (Tf), and maximum (Tmax) temperatures of the polymerization exotherm.
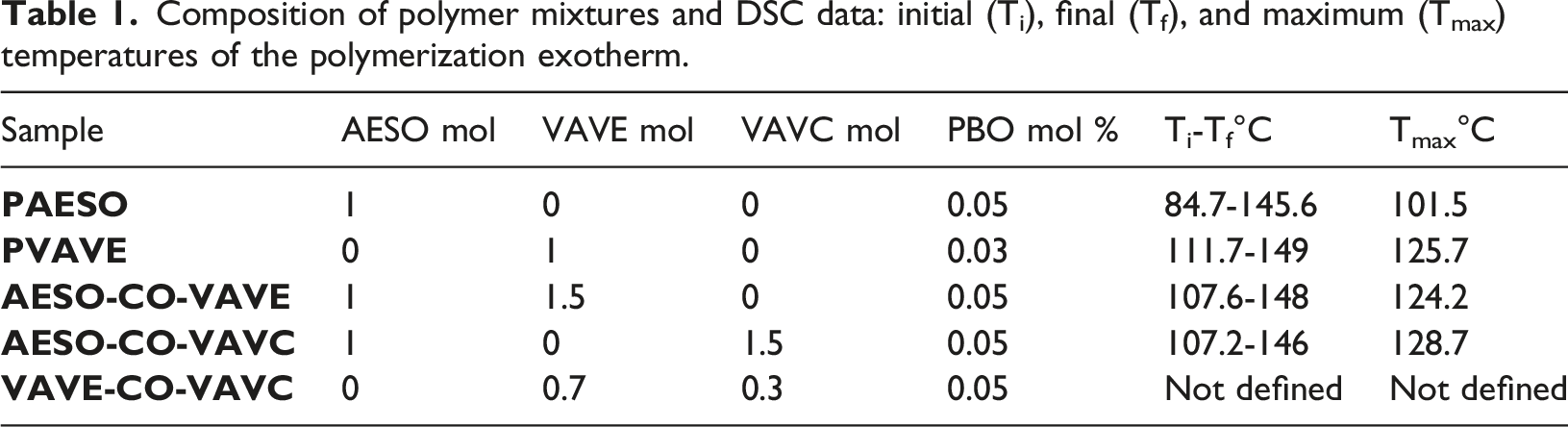
Differential Scanning Calorimetry (DSC) analyses were performed on both the individual monomers and their mixtures to obtain information regarding the polymerization exotherm. With exception of mixture
Results and discussion
Vanillin and vanillyl alcohol characterization results
Vanillyl alcohol was synthesized following previously reported procedures and characterized using FTIR-HATR, 1H NMR, 13C NMR (DEPT-135 analysis), and DSC techniques.
The FTIR spectrum of vanillyl alcohol was analyzed, with band assignments based on literature references.29,30 The characteristic carbonyl stretching vibration of the aldehyde group from vanillin at 1660 cm-1 was absent in the spectrum of vanillyl alcohol. Additionally, vanillyl alcohol exhibited a distinct characteristic band at 3445 cm-1, attributed to free hydroxyl groups. Signals associated with the methylene group in alkyl alcohols (R–CH2–OH) were detected at 1430 cm-1. Furthermore, two bands at 1033 and 995 cm-1 were observed, corresponding to the stretching vibrations of CH2–OH bonds. These spectral features confirm the conversion of vanillin to vanillyl alcohol via reduction.
Vanillyl alcohol (
The chemical structures of vanillin and vanillyl alcohol were further confirmed by 13C NMR using DEPT-135 analysis. The methine carbon adjacent to the aldehyde group in vanillin appeared at 129.05 ppm in the positive phase. Upon reduction, this carbon was converted into a methylene group in vanillyl alcohol, shifting upfield to 63.44 ppm and appearing in the negative phase, consistent with its new chemical environment as part of an alkyl alcohol.
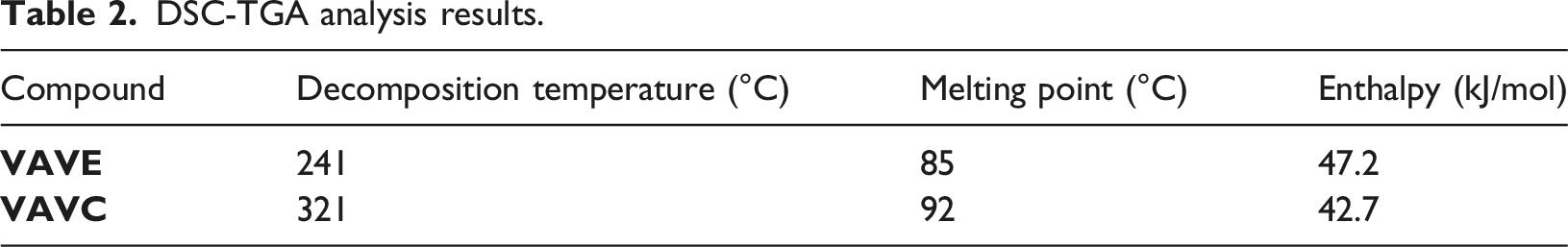
The vanillyl alcohol exhibited a melting point of 115°C, aligning with the literature range of 110–117°C, 27 and the measured enthalpy was 30.08 kJ/mol, closely matching the literature value of 30.74 kJ/mol. 32 Minor discrepancies in melting point and enthalpy may be attributed to variations in sample purity, crystallinity, or experimental conditions.
Characterization of vinylic aromatic derivatives of vanillin
The synthesis of vinylic aromatic derivatives of vanillin was carried out as previously described, and its characterization was performed using FTIR-HATR spectroscopy, 1H and 13C NMR, and thermal analysis by DSC-TGA.
Figure 2 shows the FTIR spectra of the vinylic aromatic derivatives of vanillin: FTIR spectra of vinylic aromatic derivatives of vanillin: VAVC.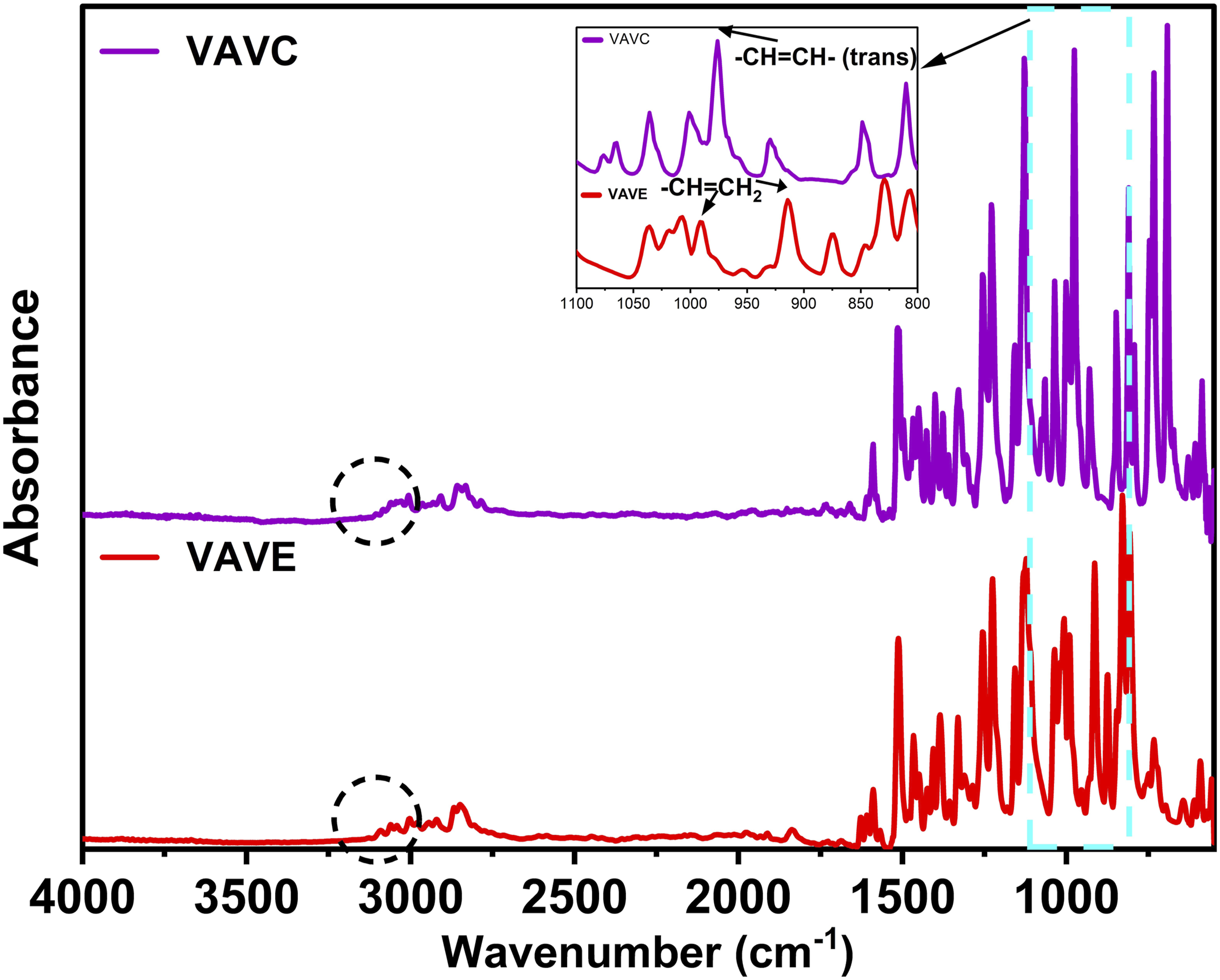
The disappearance of hydroxyl group signals in the spectra of
Characteristic signals related to the vinyl double bonds are highlighted in the spectra: a band at 975 cm-1 in the
Figure 3 presents the 1H NMR spectra of the vinylic aromatic derivatives of vanillin: 1H NMR spectra of vinylic aromatic derivatives of vanillin: 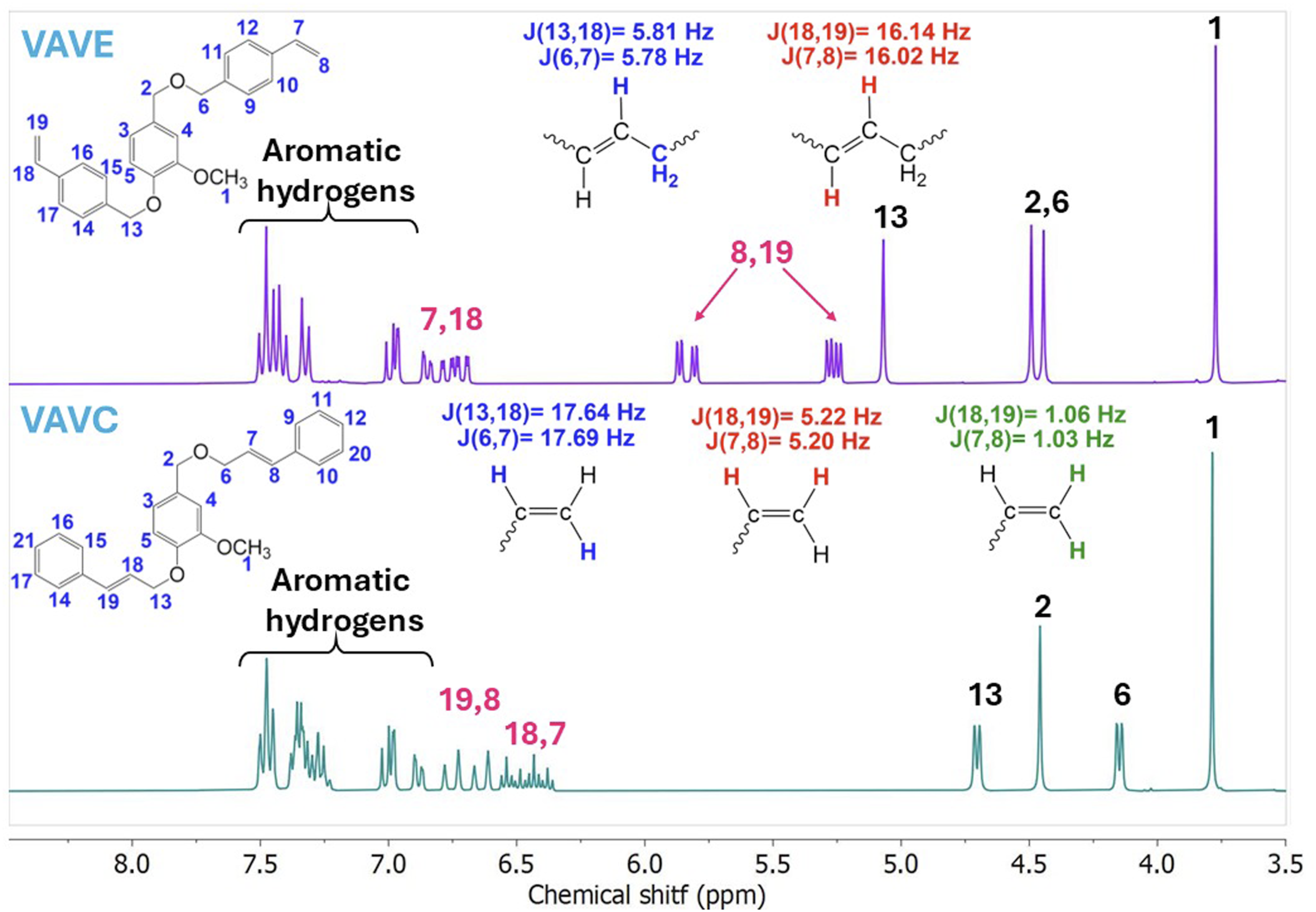
Vinylic protons in
DSC-TGA analysis results.
Characterization of polymers
Five polymers were synthesized as described in Table 1. Polymerization times varied depending on the composition. Post-curing was applied to the
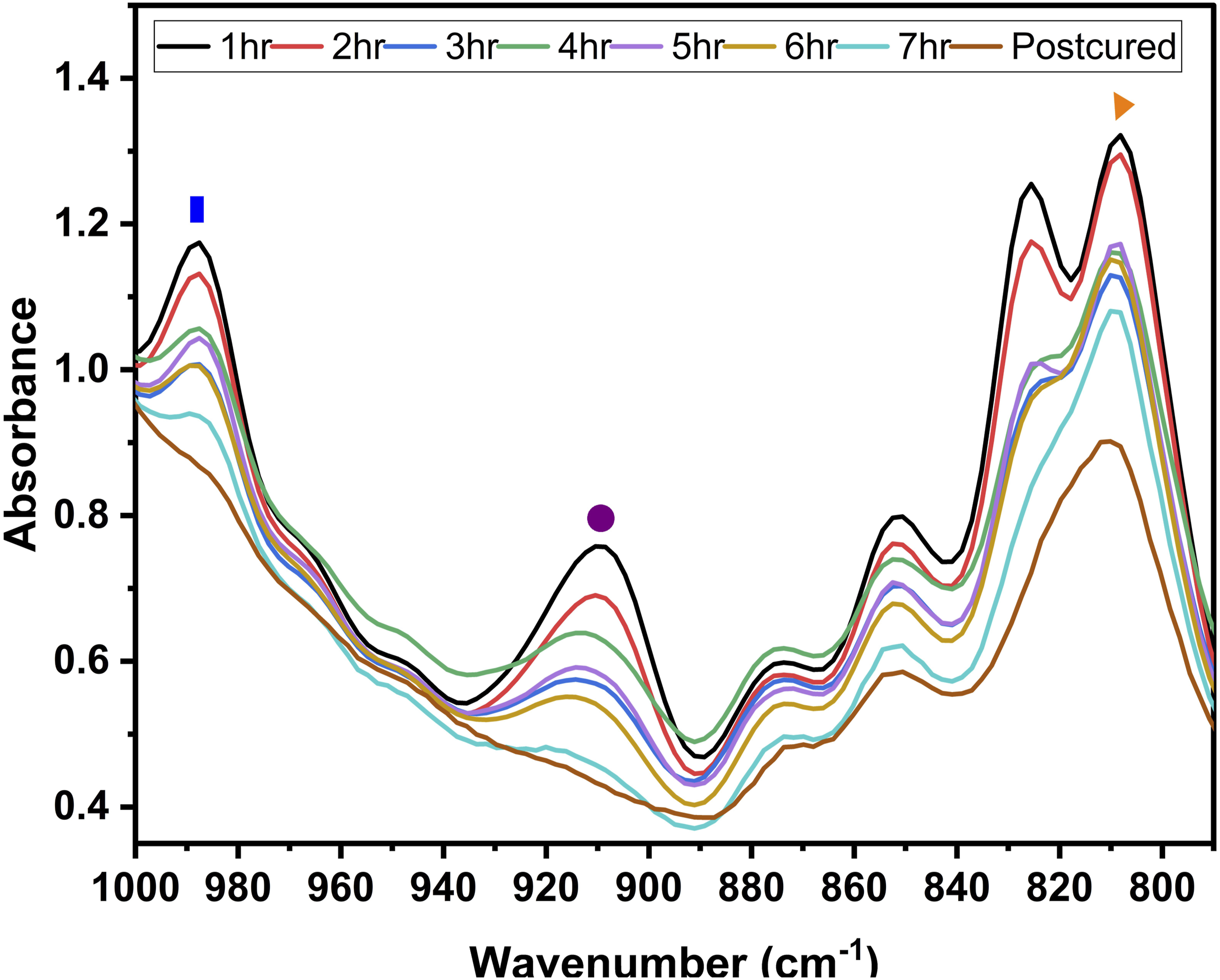
To confirm the double-bond conversion (DC (%))—HATR-FTIR spectra were collected from the reaction mixtures at the initial stage. Subsequent spectra were recorded hourly throughout the polymerization process to monitor the progression of the double-bond reaction. After normalizing the spectra, the reduction or disappearance of peak areas associated with –C = C–H stretching vibrations indicated the transformation of reactive groups into a crosslinked structure. Characteristic –C = C–H absorption bands for the polymers were observed at: 1600, 985, and 808 cm-1 for
As an example, Figure 4 illustrates the progression of DB conversion between • The signal at • The signal at • The signal at Continuous monitoring of the AESO– VAVE copolymerization reaction by FTIR.
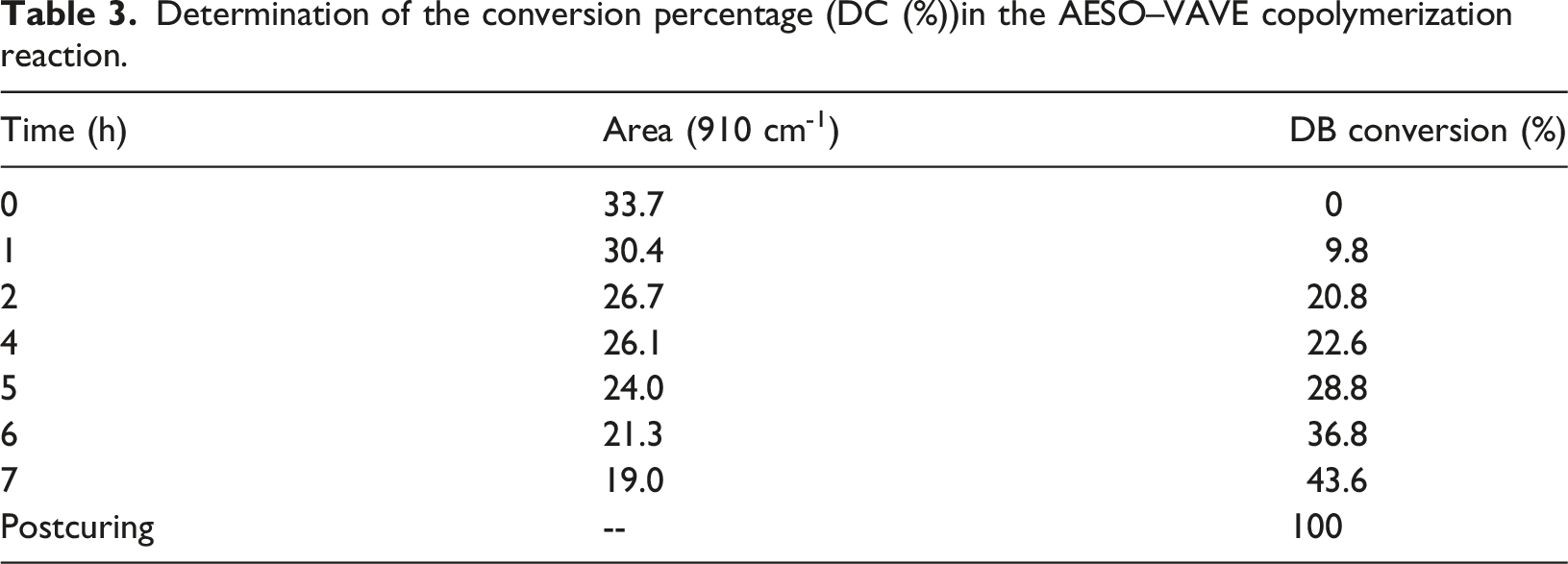
The reaction progress, DC (%) was determined by analyzing spectral changes in the 910 cm-1 band and applying equation (1).34,35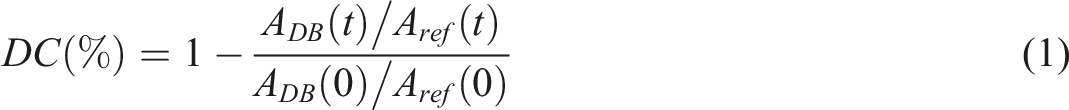
Considering that, once the curing process begins, the signals at 990 and 809 cm-1 broaden, thereby increasing their area under the curve, it is important to note that the latter signal does not disappear completely. As previously mentioned, this band represents an overlap of multiple vibrational modes. Therefore, for determining the percentage of conversion, only the area under the 910 cm-1 signal is considered.
Determination of the conversion percentage (DC (%))in the AESO–VAVE copolymerization reaction.
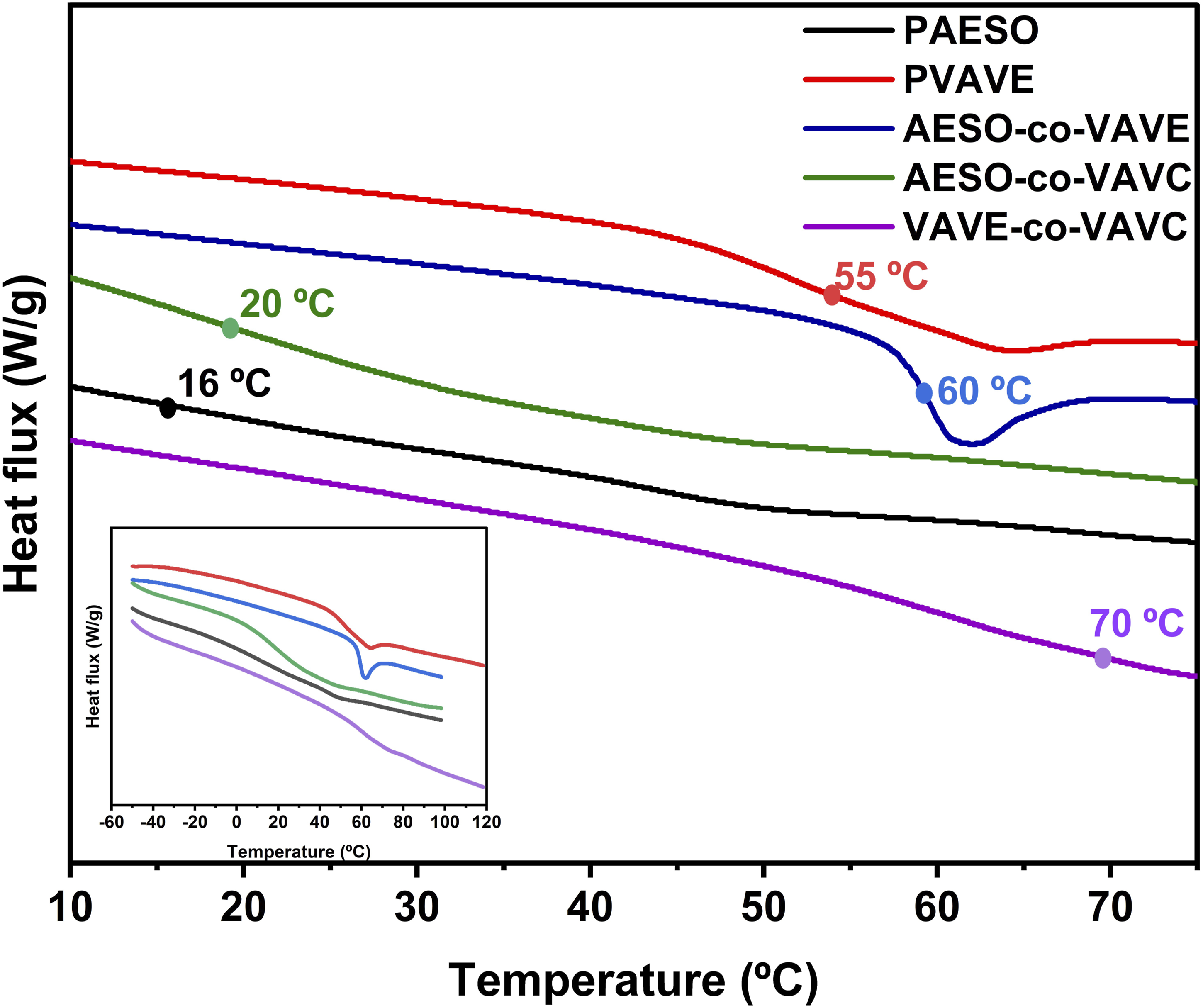
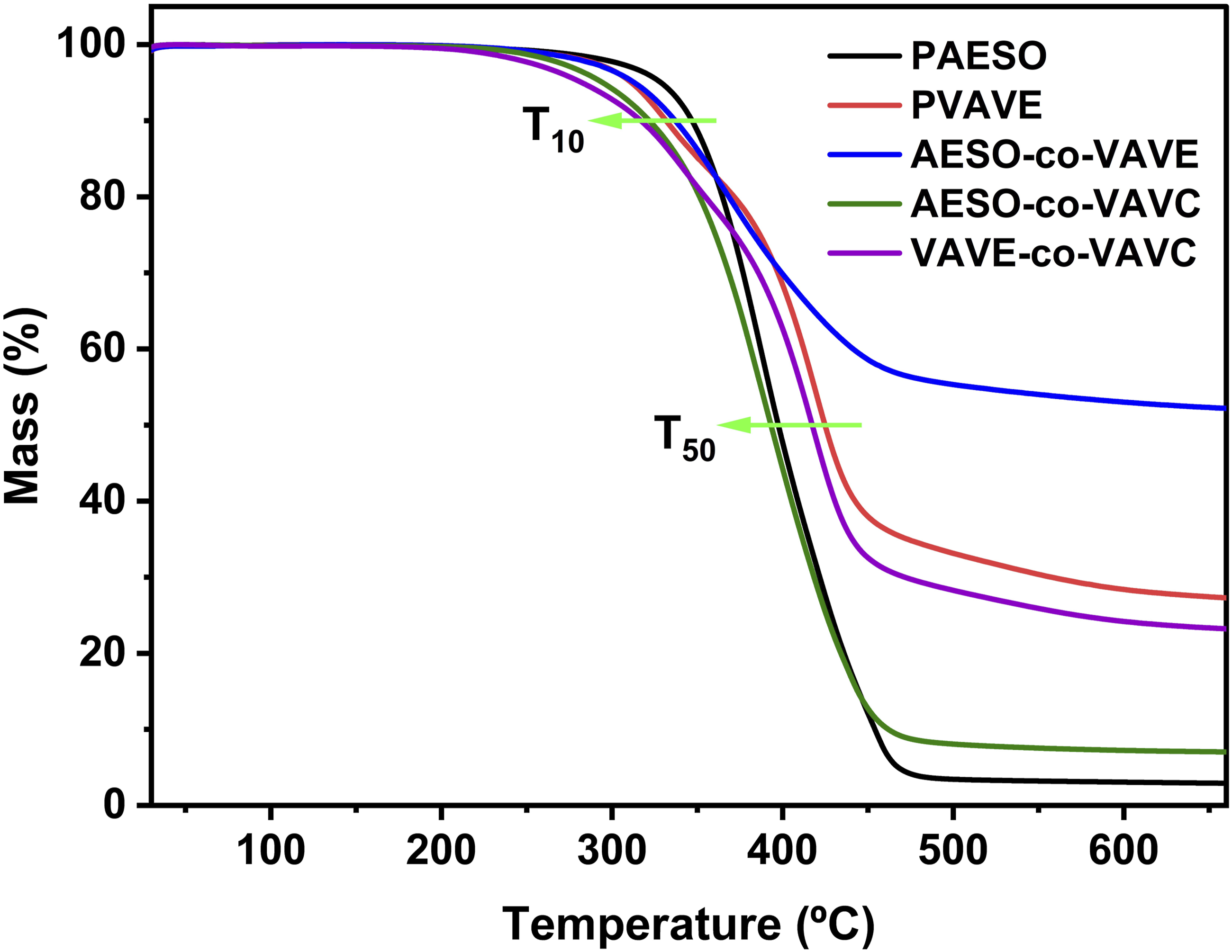
With the exception of the Progress of the crosslinking reaction for Double bond conversion, decomposition temperatures (T10 and T50), and Tg for po-lymers. Comparative Tg of the polymers obtained by DSC. TGA curves of the polymers showing T10 and T50.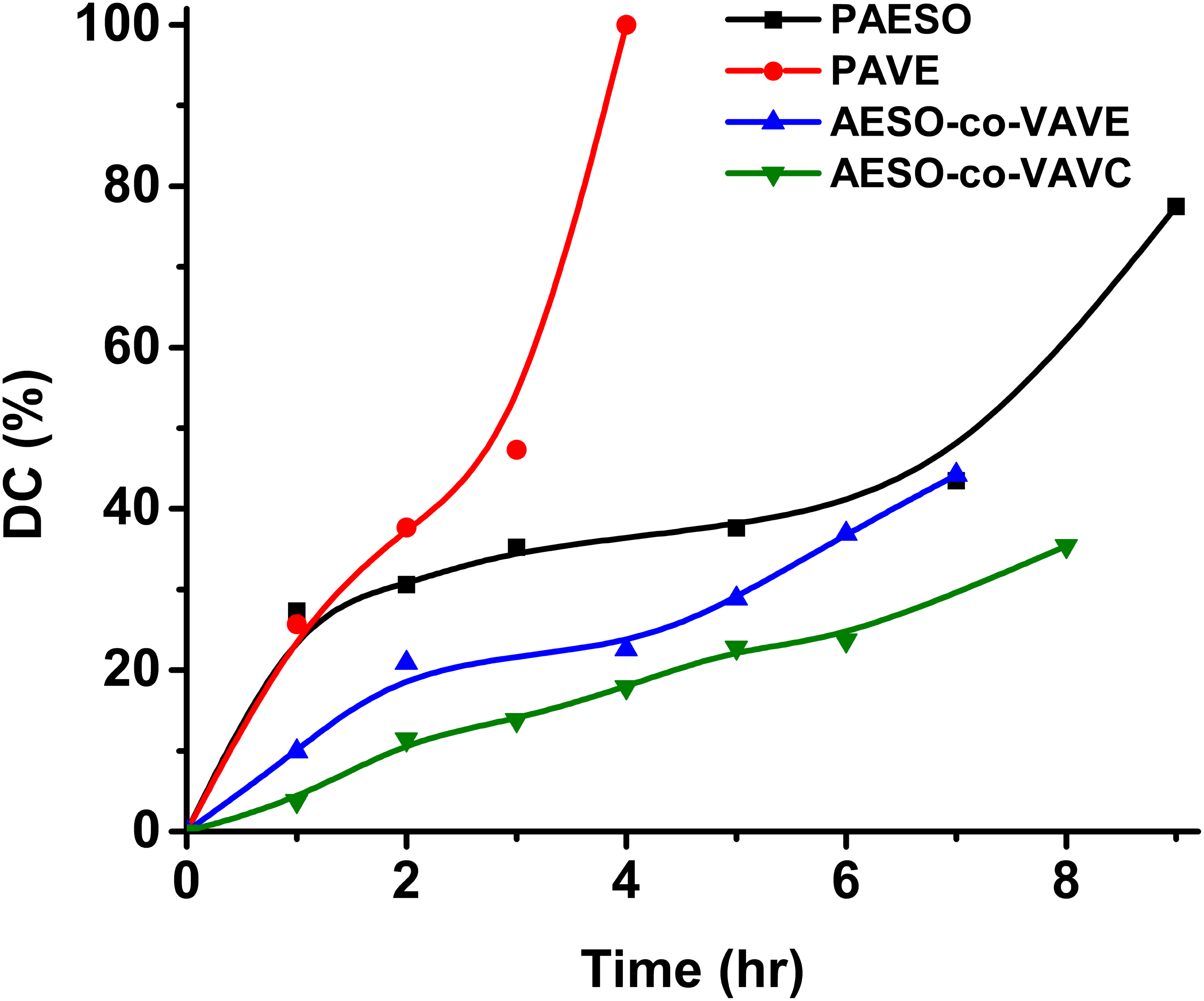
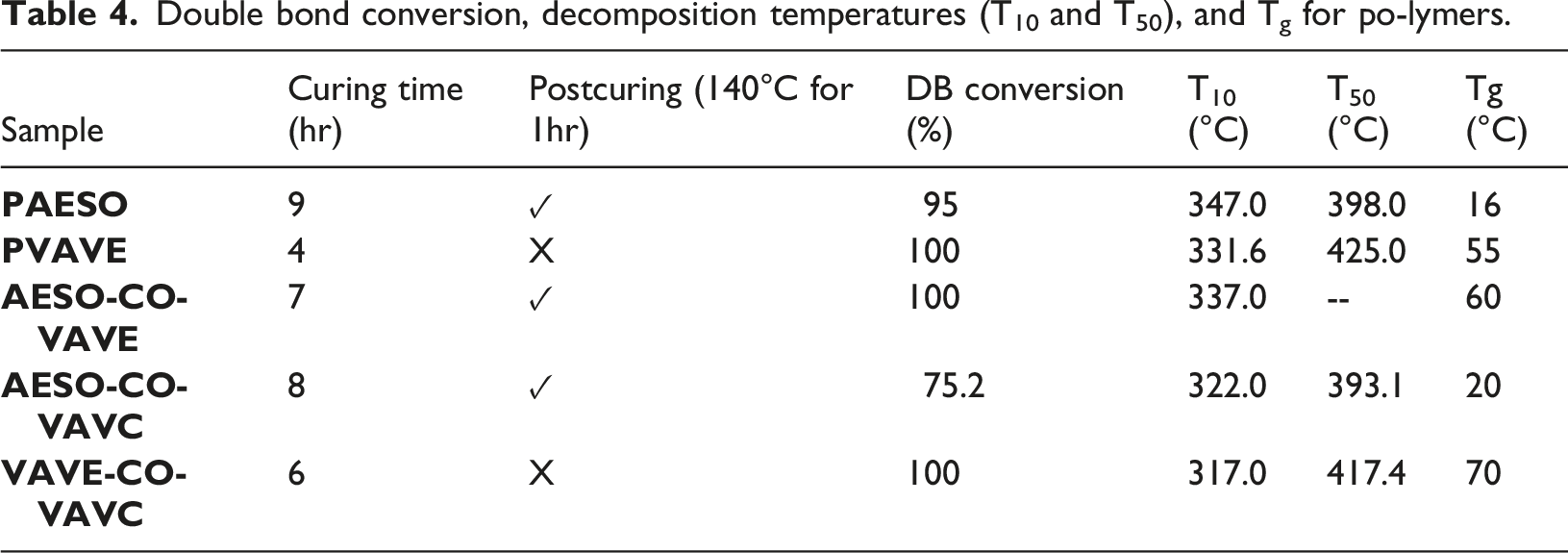
The variation in curing times observed among the different samples can be attributed to differences in polymerization kinetics and network formation, which are strongly influenced by the chemical structure of the monomers involved.
When
Overall, the curing times reflect a combination of kinetic limitations, steric effects, and vitrification phenomena. Terminal double bonds promote faster polymerization, while internal double bonds and AESO’s flexible aliphatic segments introduce delays due to reduced reactivity and increased steric hindrance. These results highlight the critical role of double bond positioning and molecular architecture in determining the curing behavior of bio-based copolymers.
The visual characteristics of the solid crosslinked polymer films are shown in Figure 8. The homopolymer Cured polymers: (1) 
The thermal degradation of all polymers was characterized by thermogravimetric analysis (TGA) under a nitrogen atmosphere, as previously described (Figure 7). Weight loss was recorded as a function of temperature, and the results are summarized in Table 4. For all polymers, thermal decomposition occurred in a single stage, within the temperature range of 315–420°C.
In contrast,
Regarding the glass transition temperature (Tg) values, this behavior is associated with the presence of aromatic rings, as polymer properties are closely linked to their chemical structure.
For homopolymers,
These results demonstrate that the incorporation of vanillin-based monomers significantly enhances the thermal stability of the resulting polymers. Similar findings have been reported in other studies, where both photopolymerization and thermal initiation led to increased rigidity and brittleness. The behavior observed in
Once the polymers were obtained,
By combining these three solvents, it is possible to establish a comparative framework: polymers that resist swelling in all three media can be considered highly crosslinked and structurally compact, whereas those that show selective swelling or mass loss reveal weaker networks or the presence of soluble fractions. This solvent triad therefore enables a more comprehensive evaluation of crosslink density and structure–property correlations than would be possible with a single solvent system.
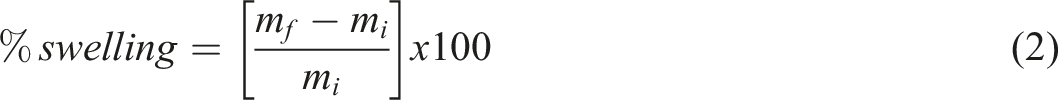
Swelling behavior of synthesized polymers in THF, toluene, and DMF.

The swelling data supports the qualitative discussion of crosslink. The swelling behavior in THF, toluene, and DMF highlights clear differences in network architecture among the polymers.
These results confirm that double bond positioning (terminal vs internal) directly influences polymerization kinetics and network architecture, and the swelling ratios provide quantitative evidence to support the structure–property correlations discussed in the manuscript.
Conclusions
This study demonstrates that the functionalization of vanillyl alcohol with 4-vinylbenzyl chloride and cinnamyl bromide provides an effective route to obtain two distinct vinylic aromatic monomers, each incorporating three aromatic rings per molecule. The resulting triple-aromatic framework represents a significant structural reinforcement compared to previously reported bio-based comonomers, which were largely aliphatic or fatty acid-derived. By introducing both terminal and internal double bonds, the monomers offer complementary advantages: terminal vinyl groups promote rapid polymerization and flexible network formation, while internal vinyl groups contribute to conjugation, efficient molecular packing, and enhanced thermal resistance.
The copolymerization of these monomers with acrylate epoxidized soybean oil (
The resulting copolymers demonstrate clear improvements in rigidity, glass transition temperature, and degradation resistance, confirming the potential of vanillin-derived vinylic aromatics as a structurally robust class of monomers. In summary, this work establishes a pathway for reinforcing vegetable oil-based resins through aromatic functionalization, and it provides a systematic evaluation of how double-bond positioning influences polymer performance. With this approach, it is now possible to envision the design of fully sustainable and bio-based synthetic routes, where hazardous reagents could be replaced by benign alternatives, thereby combining high performance with environmental responsibility.
Footnotes
Acknowledgements
The authors gratefully acknowledge SECIHTI for the Master’s fellowship. Special thanks are extended to M.C. María de las Nieves Zavala and M.C. Alejandra Núñez Pineda from CCIQS UAEM-UNAM for their valuable technical support in the 1H and 13C NMR analyses and calorimetric measurements, respectively (CCIQS SHL-2018). The authors thank SIEA-UAEMEX for the financial support for the research of this work under project No. 7217/2025CIB.
Author contributions
A.I. Miranda Urbina: Methodology (experimental work, material characterization), Software, Data Analysis, Investigation, Writing–Original Draft.
E. Vigueras Santiago: Resources, Formal analysis, and Supervision.
J.F. Nieto Alarcón: Methodology, Data Analysis and Supervision.
S. Hernández López: Project administration, Conceptualization, Formal Analysis, Writing – Review & Editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Secretaría de Investigación y Estudios Avanzados, UAEMEX; 7217/2025CIB.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.