
Abstract
This article reports on two unrelated young female patients (gender assigned female at birth) who initially presented with hypertension. Patient 1 was admitted to the hospital with an acute cerebral infarction due to poorly controlled hypertension, while Patient 2 was a first-time visitor presenting with hypertension as the initial complaint. Physical examination on admission revealed absent breast development, feminized and immature external genitalia, and no history of menarche. Laboratory investigations showed markedly elevated adrenocorticotropic hormone (ACTH) levels, significantly decreased cortisol levels and a disrupted circadian rhythm. For sex hormones, progesterone was markedly elevated, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were mildly increased, while estrogen and testosterone levels were low. Combined with karyotype analysis and gene sequencing, Patient 1 was confirmed to have a karyotype of 46,XX, and Patient 2 had a karyotype of 46,XY. In addition, pathogenic allelic variants in the CYP17A1 gene (c.985_987delinsAA) differed between the two patients: Patient 1 harbored a heterozygous mutation, whereas Patient 2 had a homozygous mutation. Both patients were ultimately diagnosed with 17α-hydroxylase deficiency (17-OHD). During the 6-month follow-up after initiation of dexamethasone replacement therapy, the patients’ blood pressure and serum potassium levels were well controlled. This case suggests that although 17-OHD is a rare disorder, clinicians should be alert to the possibility of 17-OHD in patients presenting with disorders of sex development or primary hypertension. Early and definitive diagnosis followed by appropriate glucocorticoid replacement therapy can significantly improve the patient’s quality of life.
Keywords
Introduction
17α-Hydroxylase/17,20-lyase deficiency (17OHD) is a rare variant of congenital adrenal hyperplasia (CAH), accounting for approximately 1% of all CAH cases 1 This condition results from mutations in the CYP17A1 gene, which lead to impaired activity of both 17α-hydroxylase and 17,20-lyase enzymes. Consequently, the synthesis of glucocorticoids and sex hormones is disrupted, while the mineralocorticoid pathway becomes overactivated. 2 Clinically, patients with 17OHD typically exhibit signs of mineralocorticoid excess, including hypertension, hypokalemia, and alkalosis, along with differences of sex development (DSD). Females with 46,XX karyotype often present with primary amenorrhea and lack of secondary sexual characteristics, whereas males with 46,XY sex chromosomes display diminished or lack of masculinization. 3 Owing to its rarity and heterogeneous manifestations, 17OHD is frequently misdiagnosed or overlooked. Accurate diagnosis generally requires a combination of hormonal profiling and genetic analysis. 4 Recent advances in molecular diagnostics have improved the understanding of genotype–phenotype correlations in 17OHD. Different mutation types can lead to varying degrees of enzymatic deficiency, contributing to a broad spectrum of clinical presentations. 5 In this report, we analyze two genetically confirmed cases of 17OHD and review recent literature to summarize the clinical features, diagnostic approaches, and management strategies for this disorder. Our aim is to enhance clinicians’ awareness and competence in the diagnosis and treatment of this rare disease.
Case summary
Patient 1, an 18-year-old individual raised as female, was admitted to the Department of Neurology, Hengshui People’s Hospital, on July 8, 2023, due to acute cerebral infarction. During hospitalization, she was transferred to the Endocrinology Department due to recurrent hypokalemia and poorly controlled blood pressure. Her medical history included a 4-year record of hypertension and developmental disorder. The patient reported prior evaluation at an external hospital, where chromosomal analysis reportedly indicated a 46,XX karyotype (no formal report available) and “rare disease was not excluded” (details unknown), but no treatment had been initiated. Physical examination revealed: T 36.2°C, P 92 bpm, R 18 bpm, BP 143/78 mmHg. Height 178 cm, weight 73 kg, and BMI 23.04 kg/m2. Breast development was absent. The external genitalia were female, infantile in appearance, with no history of menarche. The external genitalia were female, infantile in appearance, with no history of menarche. Hormonal evaluation showed no significant thyroid dysfunction. Adrenocorticotropic hormone (ACTH) was markedly elevated (8:00-16:00-0:00:121.692-42.335-30.370 pg/ml), and cortisol levels were significantly subnormal, with a disruption of cortisol rhythm (8:00-16:00-0:00 23.69-4.24-6.82 nmol/l). Renin (1.52 pg/ml) was suppressed, and aldosterone (64.457 pg/ml) levels were normal. Progesterone was significantly increased (11.480 ng/ml). Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) were mildly elevated (FSH 15.890 mIU/ml; LH 11.140 mIU/ml), while estrogen (E2) and testosterone (T) were low (E2 14.730 pg/ml; T 0.170 ng/ml; Table 1). Using high-throughput whole-exome sequencing (WES), a heterozygous variant in the CYP17A1 gene, designated as c.985_987delinsAA (p.Tyr329LysfsTer90), was identified in the patient (Figure 1). No other pathogenic variants were detected among the 78 core genes recommended by the American College of Medical Genetics and Genomics (ACMG). Family genetic screening revealed that the patient’s father carried the same heterozygous variant, while no pathogenic variants were detected in the patient’s mother and sister. Combined with the patient’s clinical manifestations and hormone test results, final diagnoses included the following: cerebral infarction; congenital adrenal hyperplasia (17-alpha-hydroxylase deficiency); hypertension stage 3, very high risk; electrolyte imbalance; and hypokalemia. Discharge medications consisted of aspirin enteric-coated tablets 75 mg once daily; atorvastatin 20 mg once nightly; butylphthalide soft capsules 2 capsules three times daily; dexamethasone 1 mg once daily at 8 AM; and levamlodipine besylate tablets 2.5 mg once daily. At the 1-week endocrinology follow-up, the patient was alert with no significant residual neurological deficits. Blood pressure was 130/85 mmHg, and serum potassium was 4.34 mmol/L.
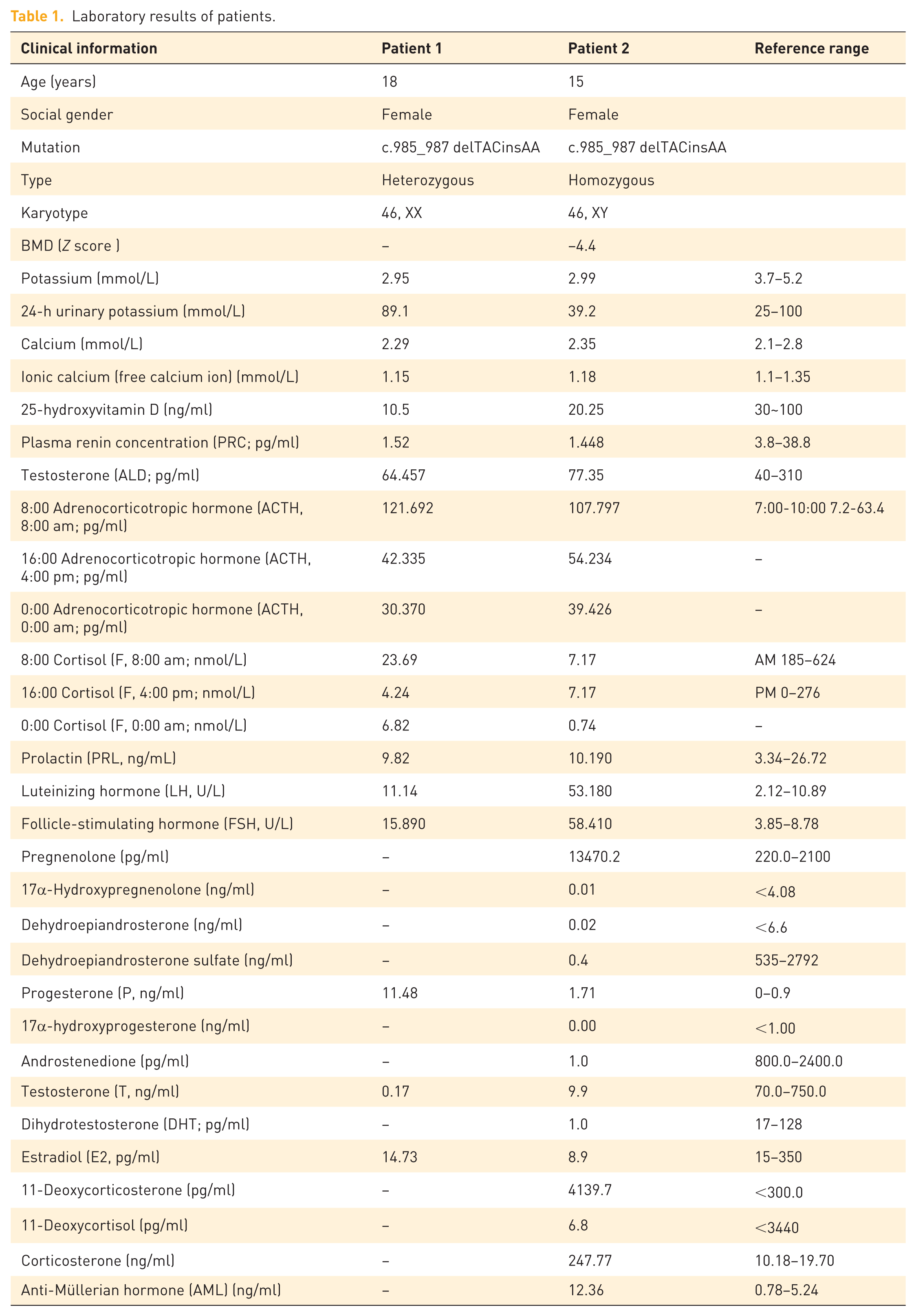
Laboratory results of patients.

Genetic test of patient 1.
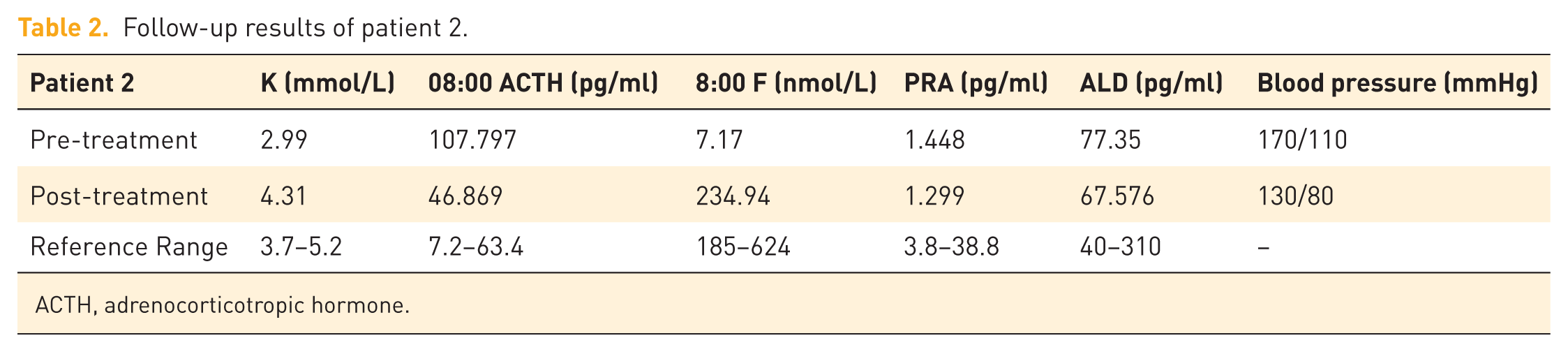
Patient 2, a 15-year-old social female, was admitted for evaluation of “developmental abnormalities for 2 years and incidentally discovered hypertension for 3 months.” Two years prior to admission, the patient presented to Hengshui Third Hospital due to primary amenorrhea. Clinical examination revealed underdeveloped breasts, and gynecologic ultrasound showed the absence of the uterus and ovaries. She was subsequently referred to the Gynecology department of our hospital for further assessment, where karyotyping identified a 46,XY chromosomal pattern. However, this finding was not further investigated at that time, and no specific diagnosis or treatment was pursued. Three months before admission, the patient was found to have elevated blood pressure during a routine physical examination, with readings as high as 170/110 mmHg. She denied symptoms such as dizziness, headache, palpitations, or muscle weakness. She self-administered nifedipine sustained-release tablets (10 mg twice daily) for blood pressure control, with suboptimal effect—blood pressure remained around 150/100 mmHg. She was then admitted to the Endocrinology department for further management. Given the clinical context, secondary hypertension was suspected, prompting hospitalization for comprehensive evaluation. The patient’s parents are non-consanguineous. There was no history of notable medication use during pregnancy. The patient was born full-term via cesarean section with a birth weight of 3.0 kg. Her growth, development, and intelligence were comparable to those of her peers. She has one younger brother and one younger sister, both developing normally and normotensive. There is no family history of similar disorders or other genetic diseases. Physical examination revealed BP 135/99 mmHg, height 173.5 cm, weight 56.5 kg, and BMI 18.8 kg/m2. She had a tall, slender build with a slightly dark complexion. Bilateral breast development was Tanner stage I. Axillary and pubic hair were absent. External genitalia were female, infantile in appearance, with no clitoromegaly or ambiguity. There was no notable hyperpigmentation of the areolae or vulva. No limb deformities were observed. Muscle strength was grade V bilaterally, with normal tone. Babinski signs were negative. Laboratory investigations showed hypokalemia with concurrent hyperkaluria, and potassium supplementation was initiated. Hormonal assays demonstrated significantly elevated ACTH (8:00-16:00-0:00 107.797-54.234-39.426 pg/ml), mineralocorticoids (11-deoxycorticosterone 4139 pg/ml; corticosterone 247.77 ng/ml), and progesterone (1.71 ng/ml), while cortisol (8:00-16:00-0:00 7.17-7.17-0.74 nmol/l) and sex hormone levels (Dehydroepiandrosterone sulfate 0.4 ng/ml; Androstenedione 1.0 pg/ml; T 9.9 pg/ml) were markedly low (Table 1). Renin (1.448 pg/ml) was suppressed, and aldosterone (77.35 pg/ml) levels were normal. Gynecologic ultrasound confirmed uterine agenesis and revealed hypoechoic masses superior to the pubic symphysis bilaterally. Inguinal ultrasonography identified bilateral hypoechoic masses within the inguinal canals, consistent with cryptorchidism. Repeat karyotyping confirmed 46,XY. Bone densitometry showed a Z-score of −4.4. Left-hand radiography indicated open epiphyses. Adrenal CT demonstrated bilateral adrenal hyperplasia (Figure 2). Genetic analysis performed by Beijing MyGenome Medical Laboratory identified a homozygous mutation in the CYP17A1 gene: c.985_987delinsAA, resulting in a frameshift mutation (p.Tyr329LysfsTer90; Figures 3). Familial segregation analysis via Sanger sequencing confirmed that both parents were heterozygous carriers of the variant. Testing of the patient’s siblings did not detect clinically significant pathogenic variants in the CYP17A1 gene (NM_000102.3). Based on the comprehensive diagnosis of congenital adrenal hyperplasia (17-alpha-hydroxylase deficiency), stage 3 hypertension (high risk), electrolyte disturbance, hypokalemia, and severe osteoporosis, the patient was initially prescribed dexamethasone 0.75 mg once daily. Considering the patient’s pre-existing severe osteoporosis and the potential for long-term glucocorticoid therapy to exacerbate bone loss, calcitriol and calcium carbonate with vitamin D were concurrently initiated: calcium carbonate with vitamin D3 tablets 1.5 g once daily, calcitriol one tablet twice daily, and nifedipine sustained-release tablets 10 mg once daily. Subsequently, the patient sought further management at a pediatric hospital in Beijing, where hydrocortisone acetate 10 mg twice daily was substituted for dexamethasone. Following this adjustment, blood pressure control deteriorated (134/97 mmHg), and hypokalemia recurred (serum potassium 3.23 mmol/L). Since the patient had reached a final height of 175 cm with no further growth expected, and due to the relatively short half-life of hydrocortisone acetate, blood pressure and hypokalemia became poorly controlled. Therapy was therefore switched back to dexamethasone 0.75 mg once daily at 8:00 AM. On this regimen, follow-up assessments showed satisfactory control of both blood pressure and potassium levels (Table 2). Three months later, the patient underwent bilateral orchiectomy at a tertiary hospital. Her mental and psychological status improved markedly, and she independently affirmed her decision to continue living as a female.
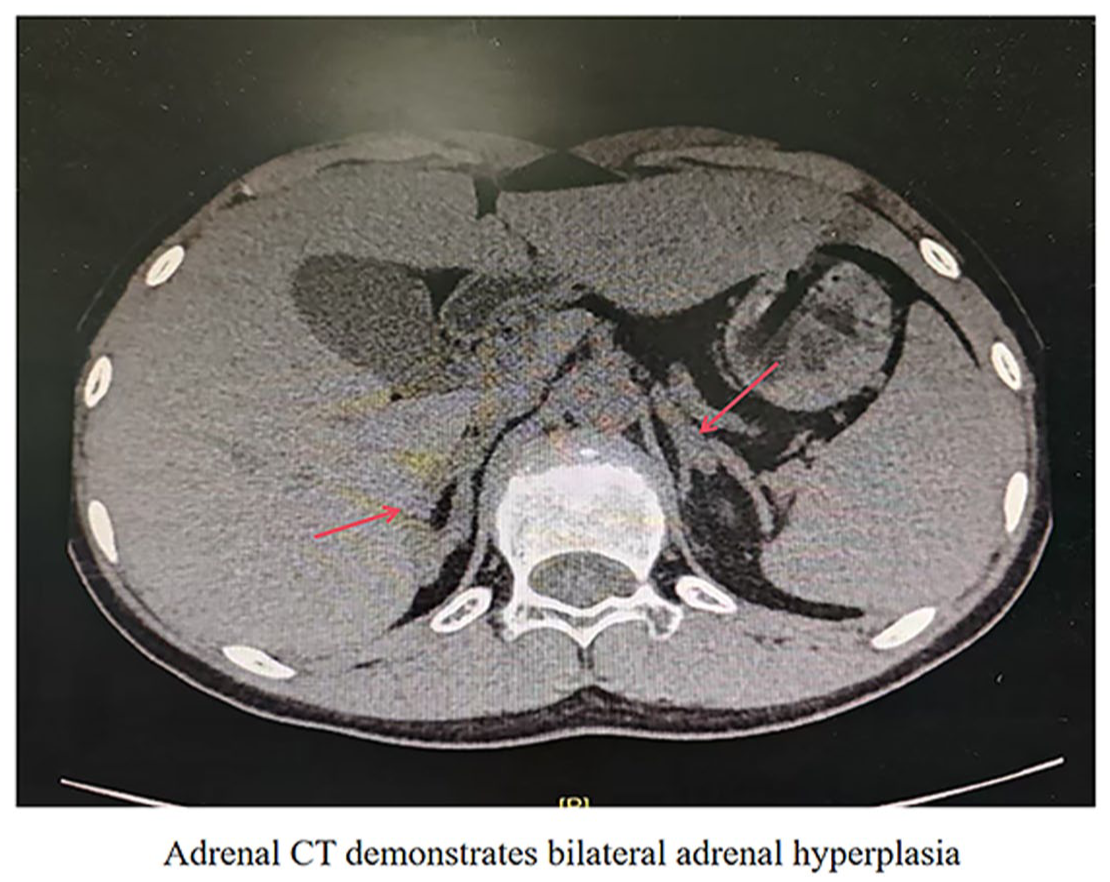
Non-contrast adrenal CT scan of patient 2.

Genetic testing of patient 2.
Follow-up results of patient 2.
ACTH, adrenocorticotropic hormone.
Discussion
Congenital adrenal hyperplasia caused by 17α-hydroxylase/17,20-lyase deficiency (17-OHD) is a rare autosomal recessive disorder. It results in impaired cortisol and sex steroid synthesis, with consequent accumulation of mineralocorticoid precursors. Patients typically present with hypertension, hypokalemia, sexual infantilism, and primary amenorrhea. 6 One consistent issue highlighted in previous reports is the variability of clinical presentation, which often leads to delayed recognition or misdiagnosis, especially when hypertension or DSD appear in isolation. 7 Our two patients reflect this clinical reality, as both experienced a prolonged diagnostic journey despite fairly typical manifestations.
Compared with other forms of congenital adrenal hyperplasia, 17-OHD tends to carry a higher cardiovascular burden. This is mainly attributed to chronic deoxycorticosterone (DOC) excess and long-standing hypokalemia. Shrikant et al. 8 reported that early-onset hypertension is common and may progress to hypertension-mediated organ damage if not adequately treated. Mattia et al. 9 further suggested that sustained DOC exposure itself can contribute to cardiovascular injury, even in young individuals. In our Patient 1, severe cerebrovascular disease developed at a relatively young age after years of uncontrolled blood pressure. Although multiple factors may have contributed, the clinical course strongly suggests the impact of long-term mineralocorticoid excess. It is worth noting that recent studies have shifted attention toward vascular injury and microvascular dysfunction as central mechanisms in hypertensive complications, rather than primary myocardial involvement. 10 The stroke observed in this patient is in line with this vascular-dominant pattern.
Endocrinologically, both patients showed the typical profile of 17-OHD, including elevated ACTH and gonadotropins with markedly suppressed cortisol, androgen, and estrogen levels. This pattern has been well documented in earlier studies.11 –13 Patient 2 had already undergone evaluation for primary amenorrhea and a 46,XY karyotype before referral, but no definitive diagnosis was made at that stage. This is not uncommon in clinical practice. Patients with DSD often move between specialties, and the diagnosis may be delayed simply because the condition sits at the intersection of several disciplines. Our experience reinforces the need for closer coordination between endocrinology, gynecology, genetics, and pediatrics.
Glucocorticoid replacement remains the cornerstone of treatment. By suppressing ACTH, it reduces DOC overproduction and helps correct hypertension and hypokalemia. In practice, however, choosing the optimal regimen is not always straightforward. Hydrocortisone is closer to physiological cortisol and is usually preferred in children, but its short half-life can make blood pressure control less stable and adherence more difficult.14,15 Dexamethasone, on the other hand, provides stronger ACTH suppression and is easier to administer once daily, though metabolic and skeletal side effects remain a concern. Both approaches have been reported in the literature. Batatinha et al. 16 described good biochemical control with low-dose dexamethasone in two 46,XY patients. Pediatric cases treated with hydrocortisone have also shown normalization of endocrine parameters and blood pressure.17,18 In our patients, dexamethasone produced a rapid and stable correction of hypertension and hypokalemia. Interestingly, when we attempted to switch Patient 2 to hydrocortisone, blood pressure and potassium control worsened, and dexamethasone had to be restarted. This suggests that in some adolescent or adult patients with more severe phenotypes, dexamethasone may offer more reliable mineralocorticoid suppression. That said, the potential long-term risks, particularly bone loss and metabolic complications, should not be underestimated. 19
Sex development and gonadal management remain complex issues in 17-OHD, and there is still no universally accepted protocol.20,21 Decisions are usually individualized, taking into account karyotype, gonadal structure, psychological identity, and social context. In 46,XY individuals, gonadectomy is generally recommended because of the theoretical risk of malignancy in dysgenetic gonads. 22 Consistent with this approach, Patient 2 underwent bilateral orchiectomy and is planned for estrogen replacement during follow-up.
Although 17α-hydroxylase deficiency (17OHD) is one of the relatively rare etiological subtypes of CAH. The incidence of 17OHD has been gradually increasing in countries such as Brazil, Japan, and China. It has now surpassed the incidence of 11β-hydroxylase deficiency, becoming the second most common type of CAH. 23 Recently, researchers 5 summarized the gene mutation types in 181 Chinese patients with 17OHD and concluded that the deletion mutation c.1459_1467del9 (p.D487_F489del) located in exon 8 and the frameshift mutation c.985_987delTACinsAA(p.Tyr329Lysfs) in exon 6 (including homozygous and heterozygous variants) are hotspot mutations in the Chinese population. Patient 2 carried a homozygous hotspot mutation, while only a heterozygous variant was identified in Patient 1, despite a very typical clinical and hormonal phenotype. Similar discrepancies between genotype and phenotype have been described before. They are often attributed to deep intronic variants or structural changes that are not captured by standard sequencing methods. This reinforces an important clinical point: genetic testing is extremely helpful, 24 but it does not replace clinical judgment, especially when the phenotype is convincing.
There are several limitations in this report. The most obvious is the small sample size, which is unavoidable given the rarity of the disease. We were also unable to perform further genetic analysis in Patient 1 due to sample constraints. In addition, long-term follow-up data are still incomplete. Despite these limitations, a few practical messages are clear. Young patients with unexplained hypertension, hypokalemia, delayed puberty, or primary amenorrhea should prompt consideration of 17-OHD early in the diagnostic process. Early recognition and timely glucocorticoid therapy are crucial to prevent irreversible cardiovascular and cerebrovascular complications. Just as importantly, management should not rely on a single specialty. A coordinated, multidisciplinary approach remains key to improving long-term outcomes.
Conclusion
Although 17-OHD is a rare disorder, clinicians should be alert to the possibility of 17-OHD in patients presenting with disorders of sex development or early-onset hypertension, particularly when associated with hypokalemia. A targeted physical examination, including pubertal assessment and genital evaluation, is essential, and delayed recognition may result in premature vascular complications. Even in cases where genetic testing reveals only a single heterozygous mutation, the combination of characteristic clinical features and confirmatory hormonal profiling is often sufficient to establish the diagnosis. Early and definitive diagnosis followed by appropriate glucocorticoid replacement therapy can significantly improve the patient’s quality of life and prevent life-threatening cardiovascular and cerebrovascular complications. This case report was prepared in accordance with the CARE guidelines. 25