
Abstract
Muscular dystrophies are inherited disorders sharing similar clinical features and dystrophic changes on muscle biopsy. Duchenne muscular dystrophy is the most common inherited muscle disease of childhood, and Becker muscular dystrophy is a milder allelic variant with a slightly lower prevalence. Myotonic dystrophy is the most frequent form in adults. Cardiac magnetic resonance is the gold standard technique for the quantification of cardiac chamber volumes and function, and also enables a characterisation of myocardial tissue. Most cardiac magnetic resonance studies in the setting of muscular dystrophy were carried out at single centres, evaluated small numbers of patients and used widely heterogeneous protocols. Even more importantly, those studies analysed more or less extensively the patterns of cardiac involvement, but usually did not try to establish the added value of cardiac magnetic resonance to standard echocardiography, the evolution of cardiac disease over time and the prognostic significance of cardiac magnetic resonance findings. As a result, the large and heterogeneous amount of information on cardiac involvement in muscular dystrophies cannot easily be translated into recommendations on the optimal use of cardiac magnetic resonance. In this review, whose targets are cardiologists and neurologists who manage patients with muscular dystrophy, we try to summarise cardiac magnetic resonance findings in patients with muscular dystrophy, and the results of studies evaluating the role of cardiac magnetic resonance as a tool for diagnosis, risk stratification and follow-up. Finally, we provide some practical recommendations about the need and timing of cardiac magnetic resonance examination for the management of patients with muscular dystrophy.
Keywords
Muscular dystrophies (MDs) are inherited disorders sharing similar clinical features and dystrophic changes on muscle biopsy. 1 Around 30 forms of MD have been identified. Duchenne muscular dystrophy (DMD) is the most common inherited muscle disease of childhood, with an estimated point prevalence in northern England of 8.29 per 100,000 boys; a milder allelic variant, Becker muscular dystrophy (BMD), has a slightly lower prevalence of 7.29 out of 100,000 boys. 1 Myotonic dystrophy (DM) is the most common form in adults, with an estimated prevalence of 10.6 out of 100,000 men, followed by facioscapulohumeral muscular dystrophy (FSHD), limb girdle muscular dystrophies (LGMDs), Emery–Dreifuss muscular dystrophy (EDMD) and other forms. 1
While respiratory failure was the main cause of death in the past, a significant improvement of respiratory assistance over the past few years has changed the natural history of the disease, and heart failure (HF) has become the leading cause of morbidity and mortality in these patients. 2 Patients with MD often develop a cardiomyopathy; the progression from a preclinical status to overt HF is usually long, and a timely diagnosis before symptom onset is crucial.
Transthoracic echocardiography has an established role as a tool for diagnosis and follow-up of MD patients, as reflected by a scientific statement from the American Heart Association (AHA). 3 Conversely, the role of cardiac magnetic resonance (CMR) is much less defined. CMR is recognised as the gold standard technique for the quantification of cardiac chamber volumes and function, including subclinical abnormalities of left ventricular (LV) kinesis that can be detected by strain imaging. CMR also enables myocardial tissue characterisation, with the detection and measurement of both diffuse myocardial fibrosis and areas of fibrotic replacement. Over the past few years, a great research effort has been made to define CMR findings in the different forms of MD, and the possible roles of CMR in the management of MD patients. Nonetheless, CMR is mentioned in just one recommendation of the AHA scientific statement: ‘It is reasonable to consider periodic use of advanced tissue imaging modalities (e.g. CMR with contrast) in the care of DMD/BMD patients for assessment of cardiac function, particularly in patients with poor acoustic windows or for assessment of myocardial fibrosis’ (class IIa, level of evidence B). 3
The present review tries to summarise CMR findings in patients with MD, and the results of studies evaluating the role of CMR as a tool for diagnosis, risk stratification and follow-up. For source selection, we searched PubMed and Scopus in December 2019 with the terms ‘muscular dystrophy’ OR ‘dystrophy’ AND ‘cardiac magnetic resonance’ OR ‘magnetic resonance’. The reference list of relevant articles was also searched; only articles published in English were included. Given the design of this work as a narrative review, no formal criteria for study selection or appraisal were enforced. Based on the available evidence and our clinical experience, we tried to provide some practical recommendations about the need and timing of CMR examination for the management of patients with MD.
Duchenne and Becker muscular dystrophies
Genetic and molecular features
DMD and BMD are X-linked recessive diseases caused by mutations in dystrophin, a component of the dystrophin–glycoprotein complex (DGC), which acts as a membrane stabiliser preventing contraction-induced damage in both the skeletal muscle and the heart. Dystrophin is completely absent in DMD, determining a more severe phenotype, and is variably reduced in BMD. 1 DMD leads to progressive muscle weakness, with loss of ambulation by the age of 12 years and the development of respiratory and cardiac failure typically by the age of 30 years. BMD usually develops in childhood, but can start in juvenile-adult age in several ways such as progressive limb girdle muscle weakness with waddling gait, myalgias and cramps with or without myoglobinuria, in association with dilated cardiomyopathy (DCM).
Some mutations in the DMD gene cause a disorder with isolated cardiac involvement called X-linked recessive dilated cardiomyopathy (XL-DCM). 4 In female DMD/BMD carriers (DMDc/BMDc), random inactivation of the X chromosome may cause a variable expression of the dystrophinopathic phenotype. 5
Cardiac phenotype
The main features of cardiac involvement in MD are reported in Table 1. Patients with DMD typically develop DCM, which contributes to short life expectancy (less than 40 years). 1 The age of onset and severity of LV dysfunction do not correlate to the degree of muscle impairment, 6 but nearly all young adult patients with DMD show imaging evidence of cardiomyopathy. Cardiac involvement is the main determinant of prognosis in BMD, and can be the first manifestation of the disease, which usually occurs between the second and fourth decade of life. 1 In both DMD and BMD, cardiac dysfunction may not be diagnosed until a late disease stage due to severely limited physical activity. This justifies the AHA recommendation to screen patients through echocardiography or other non-invasive imaging techniques. 3
Genetic background and clinical characteristics of the most common muscular dystrophies.
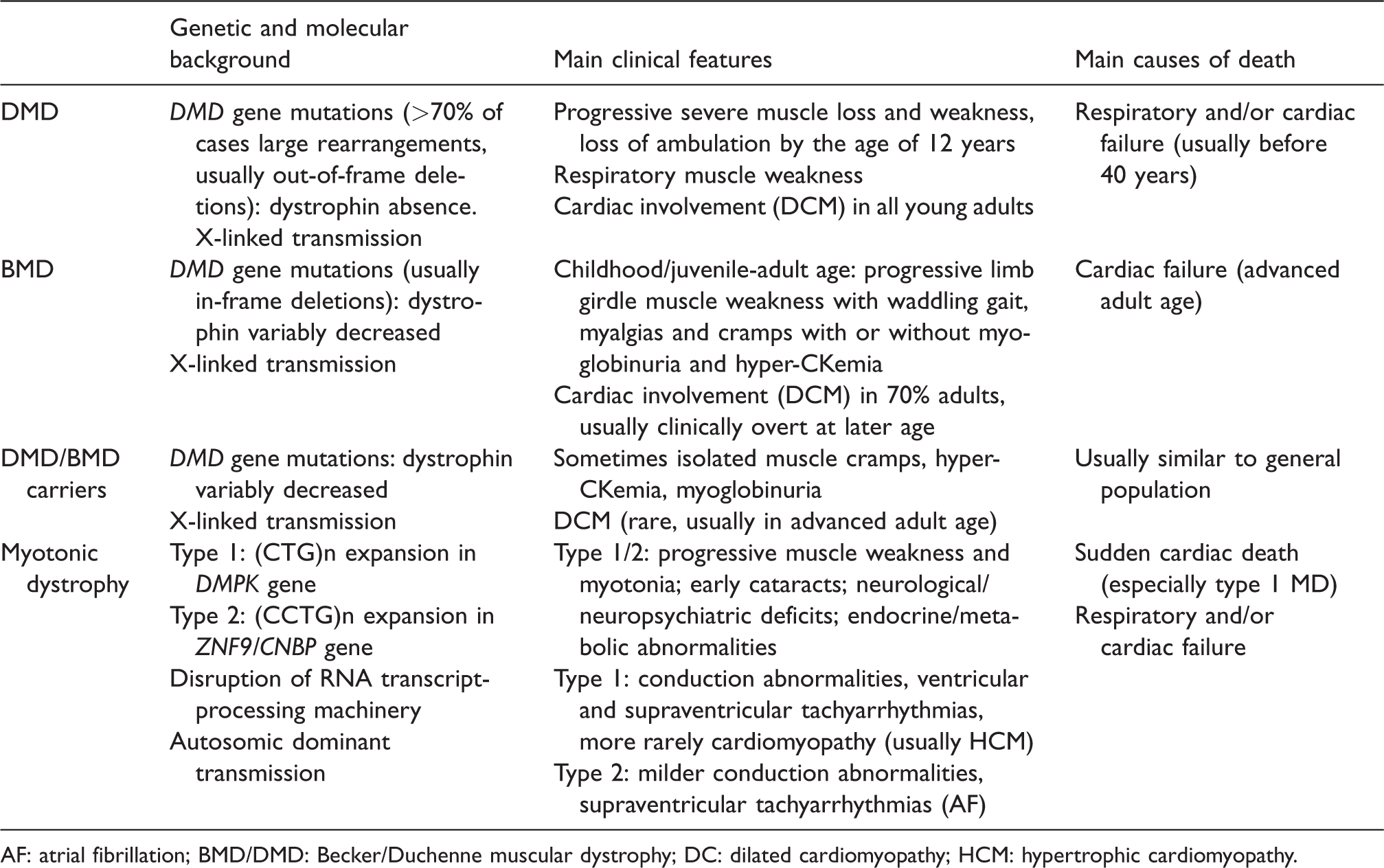
AF: atrial fibrillation; BMD/DMD: Becker/Duchenne muscular dystrophy; DC: dilated cardiomyopathy; HCM: hypertrophic cardiomyopathy.
Detection and characterisation of cardiac involvement: DMD
The role of CMR in patients with DMD is emphasised by the fact that, among patients with DMD, as many as 77% of echocardiographic studies were deemed of suboptimal quality because of a limited acoustic window. In addition, echocardiographic measures of LV function significantly over or underestimated LV function compared to CMR in 26% of cases, 7 at least partly because of suboptimal delineation of endocardial borders by echocardiography. 8
Myocardial strain abnormalities are prevalent in young DMD patients despite normal left ventricular ejection fraction (LVEF), and these strain values continue to decline with advancing age, to become even lower in patients with reduced LVEF and with late gadolinium enhancement (LGE); Hor and colleagues demonstrated a high correlation between global circumferential strain computed with tagging and feature tracking in a large cohort of DMD patients.9–12 Regional wall motion abnormalities (WMAs) often start from the basal inferolateral segment, 7 and then become more diffuse. Systolic dysfunction then develops, accompanied by LV dilation, 13 and a progressive increase in the extent of LGE. Indeed, the prevalence of LGE has been reported to increase in parallel with age and systolic dysfunction, with an apparent progression from the lateral wall to the septum that has been consistently reported. 14 Native15,16 and post-contrast17,18 T1-mapping analysis are more sensitive than LGE evaluation in detecting an expansion of extracellular spaces. For example, DMD patients with normal LVEF and no LGE displayed higher mean native T1 values than controls. 16
Most studies on DMD cohorts did not provide any specific information on right ventricular (RV) volumes and function. In a large DMD cohort of 272 patients, Mehmood et al. observed that RV systolic volumes and function were preserved and did not correlate with different categories of LV dysfunction. 18 On the other hand, the same authors described that RV volumes and function related to different categories of percentage forced vital capacity, 19 which suggests that worsening respiratory function may ultimately have a detrimental impact on the right ventricle.
Detection and characterisation of cardiac involvement: BMD
CMR is more sensitive than echocardiography in detecting systolic dysfunction and regional WMA. CMR also allows us to detect LGE, whose extent increases with age, and is significantly higher in patients with systolic dysfunction. 20 In a study carried out on 27 patients with BMD, 74% displayed LGE, most often in the basal inferolateral segment (90%), followed by the basal anterolateral segment (60%) 21 (Figure 1). Extracellular volume (ECV) was increased both in segments with LGE and in those with no LGE, and global ECV correlated with the number of LGE-positive segments. We may then conclude that ECV increase is followed by the development of LGE, and that LGE expands in parallel with deteriorating systolic function. 21
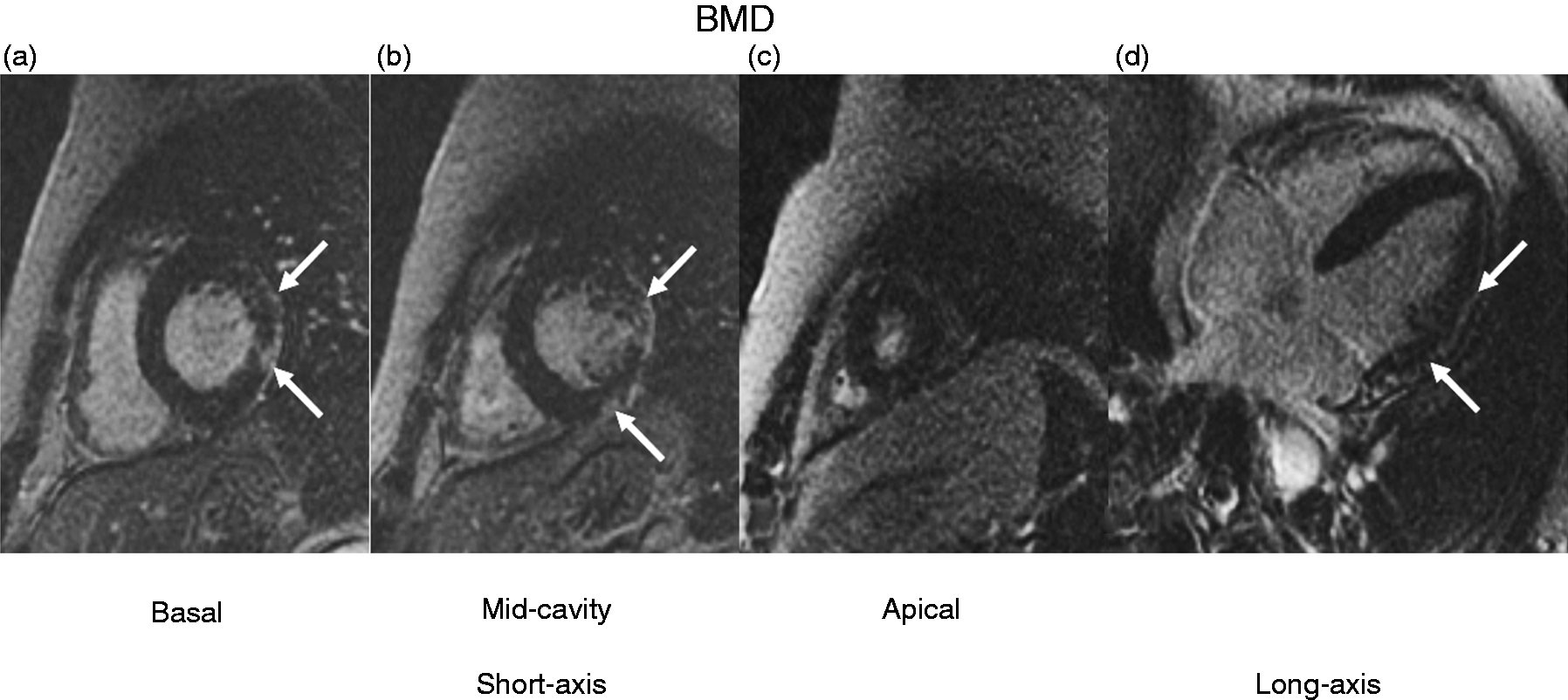
Typical late gadolinium enhancement pattern in Becker muscular dystrophy (BMD).
A more in-depth evaluation of LV mechanics reveals a reduction in global circumferential strain, and an increase in the dyssynchrony index values during the whole cardiac cycle and during diastole. 22 LGE was found in 88% of patients, usually in the lateral wall; its location was most commonly subepicardial (55%). Native T1, T2 and ECV values were higher in patients with BMD and highly correlated among them and with circumferential strain, global dyssynchrony index, and N-terminal fraction of pro-B-type natriuretic peptide. 22
Risk prediction and follow-up evaluation
A limited number of studies have combined the baseline assessment of patients with DMD or BMD with follow-up data, or have performed serial CMR examinations. In a cohort of 32 patients with DMD, LGE was present in 78% of patients, who were older, had a lower LVEF and higher LV volumes, as well as a higher incidence of ventricular tachycardia on ECG Holter monitoring. Nineteen patients died during the follow-up. LGE involving both the septum and the lateral free wall predicted death independent of age, LVEF and ventricular arrhythmias at baseline. 23 In other patient cohorts, LGE presence was associated with a more rapid decline of LVEF, 24 and higher all-cause mortality. 25 Furthermore, a full-thickness LGE pattern and systolic dysfunction (LVEF ≤45%) were reported to be strong and independent predictors of all-cause/cardiac death or cardiac transplantation in a cohort of patients with DMD or BMD. 26 Finally, changes of LV circumferential strain at mid-cavity level27,28 and LGE extent29,30 have been assessed as surrogate endpoints of clinical trials, being considered as indicators of disease progression.
CMR in female carriers of DMD/BMD
Detection and characterisation of cardiac involvement
Although female carriers of dystrophin mutations (DMDc/BMDc) are mostly free of skeletal muscle symptoms, they may develop a cardiomyopathy phenotype ranging from regional WMA to progressive HF, sometimes even requiring heart transplantation. 1 CMR can characterise the full spectrum of cardiac disease in dystrophinopathy carriers (Figure 2).
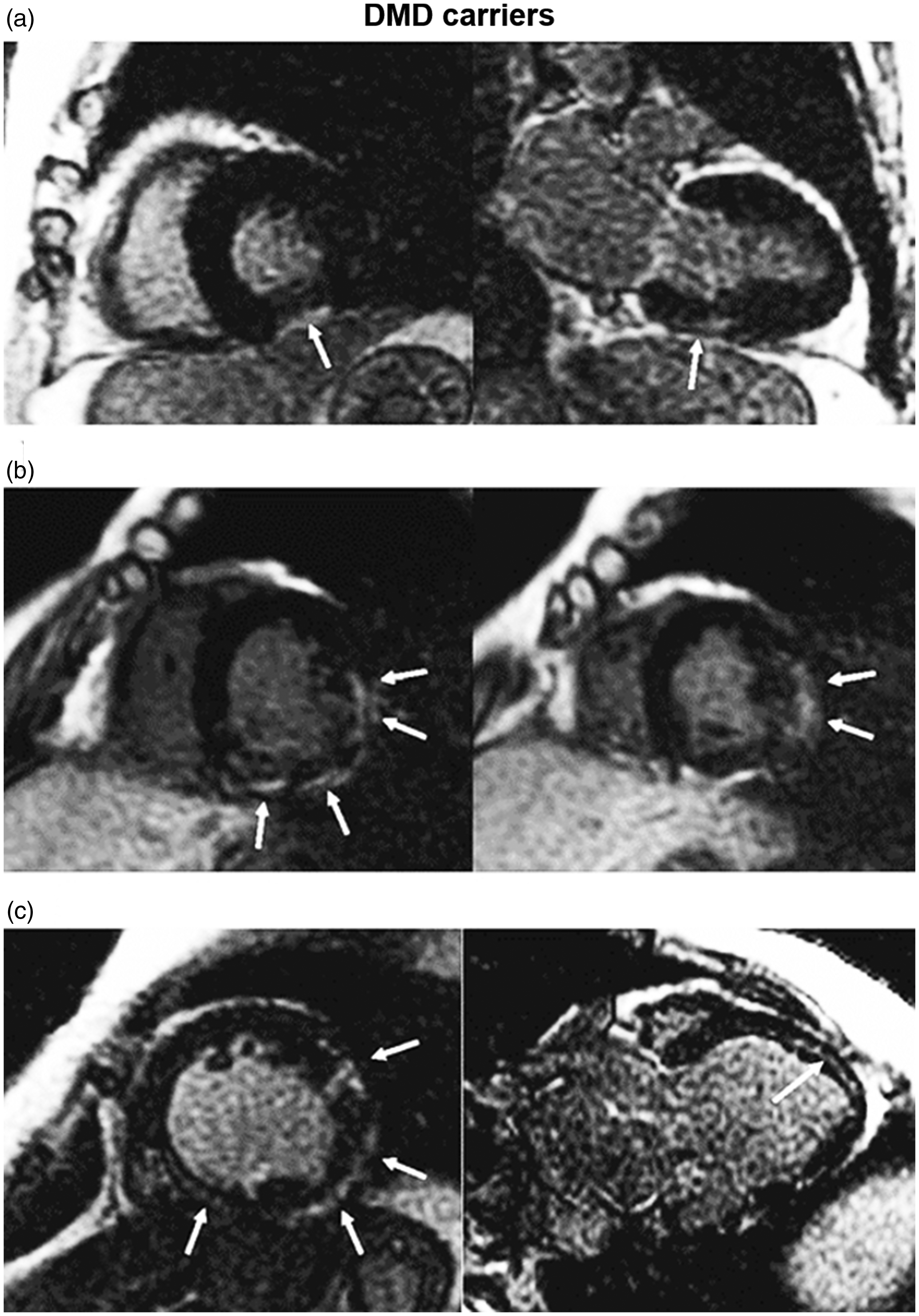
Late gadolinium enhancement (LGE) patterns in three Duchenne muscular dystrophy (DMD) carriers. (a) Focal LGE (arrows) confined to the subepicardial layer of the basal inferior segment. (b) Large area of subepicardial LGE, observed in basal and mid segments of the inferior, inferolateral and anterolateral walls. (c) Extensive LGE detected in the mid-wall layer of the interventricular septum and in the subepicardial layer of the left ventricular lateral wall.
Among 20 DMDc and 16 BMDc subjects, 47% displayed at least one pathological finding, with reduced LVEF in 14% and LGE in 44%. LGE had a median extent of 4.9% of the LV mass, and was constantly present in patients with LVEF less than 55%. LGE had a subepicardial pattern in 88% and an isolated mid-wall location in 6%. The basal inferolateral wall was most frequently involved (88%), followed by the basal anterolateral wall (38%). DMDc had significantly higher LV end-systolic volumes, lower LV and RV systolic function, higher LGE presence and extent. Interestingly, among mother–daughter couples, LGE displayed similar patterns, but its extent was almost double in mothers. Male patients displayed the same LGE patterns as their respective relatives, but with a significantly higher prevalence of cardiac abnormalities, and none of the female carrier relatives of male patients with normal CMR studies had any pathological CMR finding. Finally, the DMDc status and high serum creatinine kinase (CK) were predictive of pathological CMR findings. 31
The association between higher CK levels and LGE, which was also reported in another study on 20 female DMDc, 32 suggests a parallel deterioration of the cardiac and skeletal muscle. Notably, in a study on 30 DMDc absolute LGE and LGE percentages were higher in patients with muscle weakness than in those without this symptom. 33 Conversely, no relationship between CMR findings and age or type of mutation has been reported. 32
Limited evidence exists on cardiac energy metabolism assessment in patients with DMD/BMD or mutation carriers. A small study compared cardiac energetics in 13 BMD, 10 female carriers and 23 controls by means of phosphorus-31 magnetic resonance spectroscopy and echocardiography. The cardiac phosphocreatine to adenosine triphosphate ratio was significantly reduced in patients and carriers compared with controls (P<0.001) and did not correlate with LVEF. 34 A deranged energy metabolism thus seems to represent an early manifestation of cardiac involvement, and possibly a mechanism linking altered dystrophin expression to the development of cardiomyopathy.
Risk prediction and follow-up evaluation
Very few data are available on the evolution of CMR findings over time in DMDc/BMDc. Among seven DMDc, one subject with systolic dysfunction and diffuse LGE developed severe HF and died 6 months after CMR despite medical treatment. 35 In another series, only eight DMDc out of 20 underwent a follow-up CMR examination after 12 months; none of the seven subjects without LGE at baseline developed it, while LGE extent, ECV and LV function remained substantially unchanged in the remaining subject. 32
Myotonic dystrophy
Genetic and molecular features
DM is an autosomal dominant disorder characterised by difficult relaxation of muscles after contraction, progressive skeletal muscle wasting and increased susceptibility to arrhythmias.3,36 Two variants exist, type 1 (Steinert disease) and type 2 (Ricker disease). Type 1 DM derives from abnormal expansion of the unstable CTG repeat in the gene encoding for a protein kinase, while type 2 DM results from repeat expansion in the gene encoding for a regulatory protein. 1
In both cases, CUG/CCUG-containing transcripts accumulate in the cell nuclei forming ribonuclear inclusions which cause the sequestration and deregulation of several RNA binding factors, including splicing regulators. Indeed, some of the classic manifestations of DM-like myotonia, insulin resistance and cardiac involvement can be partly ascribed to the disruption of the alternative splicing of the muscle chloride channel ClC-1, of the insulin receptor and of the cardiac troponin T, respectively. Other pathogenic mechanisms are the intracellular accumulation of RNA transcripts containing expanded triplets and the dysregulation of various micro-RNA. For example, mir-1, a micro-RNA whose deregulation has been associated with myocardial hypertrophy, ischaemia and arrhythmias, seems to be significantly reduced in the myocardial tissue of DM patients. 37
Cardiac phenotype
Cardiac involvement is very common in type 1 DM (up to 90% of patients), and manifests with conduction abnormalities (atrioventricular and intraventricular conduction defects) and tachyarrhythmias (both ventricular and supraventricular), with a high risk of sudden cardiac death (SCD). 38 Conduction abnormalities and arrhythmias probably result from myocyte hypertrophy and focal fibro-fatty infiltration along the conduction system causing delayed myocardial activation and favouring re-entry phenomena. 36 Some degree of structural heart disease including LV dilation or hypertrophy can be found in about 20% of patients, but less than 2% present with HF symptoms. 39 In type 2 DM, cardiac manifestations are less frequent (10–20%) and usually milder, mainly represented by first-degree atrioventricular or bundle–branch block and atrial fibrillation. 40 LV dysfunction and SCD have been rarely reported.40,41
CMR in DM
Detection and characterisation of cardiac involvement
CMR studies on DM have reported highly heterogeneous results regarding the prevalence and patterns of cardiac involvement. In a study carried out on 80 patients with type 1 DM, LV systolic dysfunction was found in 25%, regional hypokinesia in 14%, LV dilation in 9% and LV hypertrophy in 8%. 42 Cardiac involvement was associated with age, male sex and ECG abnormalities, but not with disease duration, severity of neuromuscular symptoms or number of CTG repeats. Patients tended to have a reduced LV mass, as also confirmed in other studies,43,44 possibly because of reduced metabolic demands due to muscle wasting and reduced physical activity.
The estimated prevalence of LGE in patients with DM ranges from 0% 43 to 42%, 45 and there is no agreement among different studies on the most common pattern of distribution of LGE.42,45–47 LGE seems to develop before systolic dysfunction, 47 but it has no association with ECG abnormalities. 46 For example, in a cohort of 52 patients with type 1 disease, a high prevalence of LGE was detected in subjects with and without alterations at 12-lead ECG warranting pacemaker implantation, with no significant difference between the two subgroups (42% vs. 43%, P = 0.999). 45 Similarly, LGE mass and LGE percentage were similar, while ECV values (albeit calculated only in the mid-ventricular septum) tended to be higher among patients with conduction disturbances (26 ± 3 vs. 24 ± 3, P = 0.050). No SCDs or major cardiovascular events occurred over one year. 45
Another study searching for earlier signs of cardiac involvement evaluated 35 patients with DM (13 with type 1 and 22 with type 2 disease), who showed lower LVEF values than healthy controls (although still in the normal range: 60 ± 8%), abnormal myocardial strain and increased native T1 relaxation time and ECV, probably due to myocardial fibrosis. 48 Non-ischaemic LGE has been characterised as the only independent predictor of atrial fibrillation/flutter on long-term ECG monitoring, 49 and signs of RV involvement (fibro-fatty replacement, myocardial thinning, or WMA) have been associated with inducible arrhythmias on electrophysiological testing. 50
Risk prediction and follow-up evaluation
CMR can be useful to detect structural and functional myocardial abnormalities in patients with DM, including early-stage disease, but the relationship between these abnormalities and conduction disturbances, the future occurrence of SCD, and also LV dilation or dysfunction remains elusive.
CMR findings in other dystrophies
A small number of CMR studies have examined patients with other MDs, due to the rarity of these disorders, the low frequency of cardiac involvement in some forms, particularly in FSHD, 51 or the frequent need for pacemaker implantation in other conditions, especially in the more advanced stages.
FSHD type 1 has an autosomal dominant transmission pattern and is the third most common inherited muscle disease. In a cohort of 52 patients, LGE was found in 25% of patients, most commonly in the basal inferolateral, inferior and septal segments. Global native T1 and ECV values were higher than controls, and segmental values were higher than controls both in LGE-positive and LGE-negative segments (suggesting diffuse interstitial fibrosis). Areas of fibro-fatty replacement were found in 13% of patients, always in the apical septum, and T2 signal in the basal and mid-cavity slices was similar to controls. Arrhythmias on ECG Holter monitoring were recorded in 23% of patients, without any clear relationship with CMR findings. 52
LGMDs are a group of seven autosomal dominant and 14 autosomal recessive disorders. In the largest cohort evaluated so far (n = 16), 15 patients (94%) had normal LV volumes, systolic function (LVEF 60 ± 7%), and systolic circumferential strain, but areas of LGE were present in 47% of cases, with subepicardial or mid-wall location. 53 No preferential LGE location was reported, while other studies have reported a preferential location in the subepicardial region of the inferolateral wall. 54 The loss of torsion with preservation of circumferential and longitudinal strain was proposed as a hallmark of LGMD 2I, 55 but the possibility to draw this conclusion is limited by the lack of a comprehensive assessment of wall motion in most other MDs.
EDMD includes a form with X-linked transmission and another with autosomal dominant transmission. In a cohort of eight patients with the second form, the only abnormality detected was a decrease in systolic circumferential strain in the inferior segment. No patient displayed areas of LGE, possibly suggesting a different pathogenesis of cardiac involvement in EDMD compared to DMD/BMD, in which fibrosis typically precedes systolic dysfunction. 56
Limitations of CMR in MDs
Although CMR imaging does not suffer from the limitation of poor acoustic windows related to body habitus or lung disease, several factors may limit its applicability in the setting of MD. 3 CMR might not be feasible for patients who are unable to be comfortably positioned because of immobility or contractures. In addition, CMR utility can be limited in patients with tachycardia, arrhythmias, high respiratory rates, or inability to remain still; sedation or anaesthesia are often not applicable because of impaired cardiac and respiratory function. Many patients can have non-magnetic resonance compatible devices, or magnetic resonance-compatible devices might not yield a good image quality. Finally, there is the potential for artefact from rods used in the treatment of scoliosis in some MD patients.
How CMR can implement the care of patients with MD
The main CMR features of cardiomyopathy related to MD are summarised in Table 2. Most CMR studies were carried out at single centres, evaluated small numbers of patients and used widely heterogeneous protocols. An even greater limitation is represented by the descriptive nature of most studies, which analysed more or less extensively the patterns of cardiac involvement, but usually did not try to establish the added value of CMR to standard echocardiography, the evolution of cardiac disease over time and the prognostic meaning of the observed abnormalities of cardiac structure and function. The result is a large amount of information about cardiac involvement in MDs that cannot be translated into recommendations on the optimal use of CMR. 3 On the other hand, we can identify several possible applications of CMR in the setting of MD, which could help clinicians to refer patients to CMR examination and might inform future clinical studies (Figure 3).
Cardiac magnetic resonance features of cardiomyopathy related to the most common muscular dystrophies.
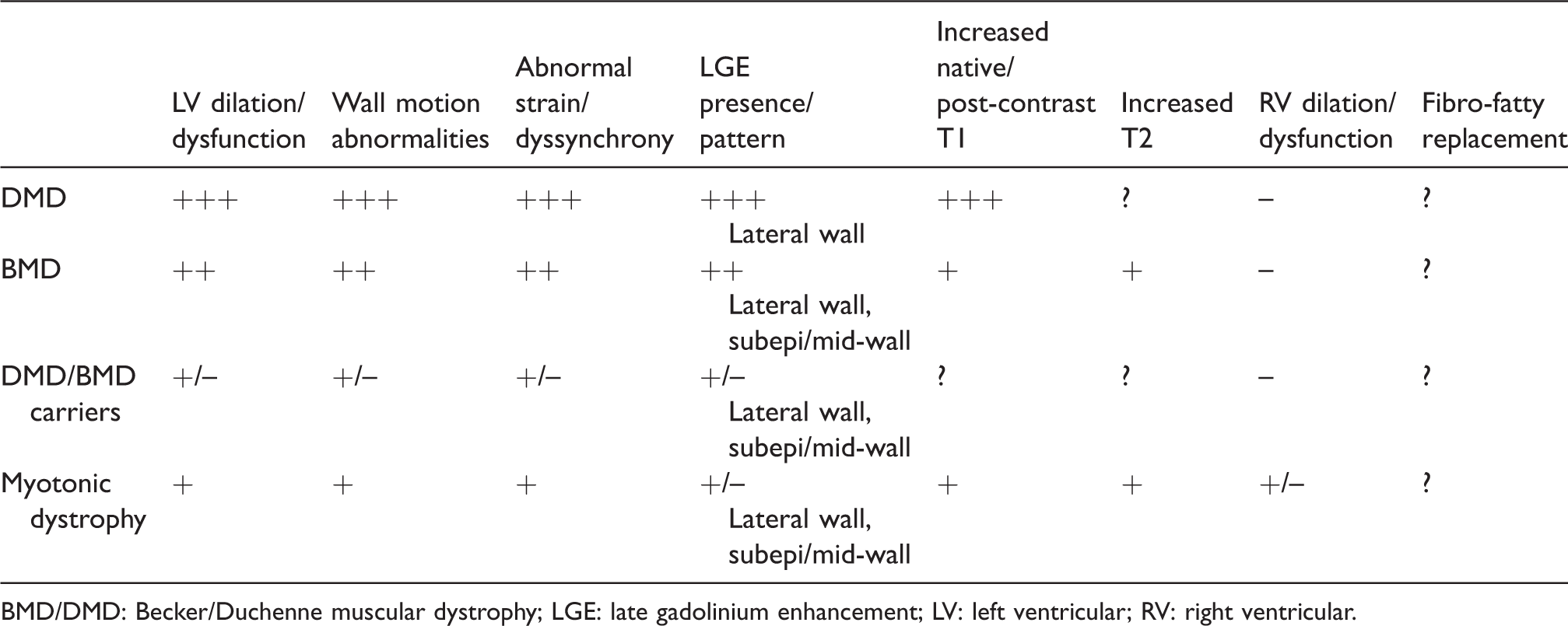
BMD/DMD: Becker/Duchenne muscular dystrophy; LGE: late gadolinium enhancement; LV: left ventricular; RV: right ventricular.
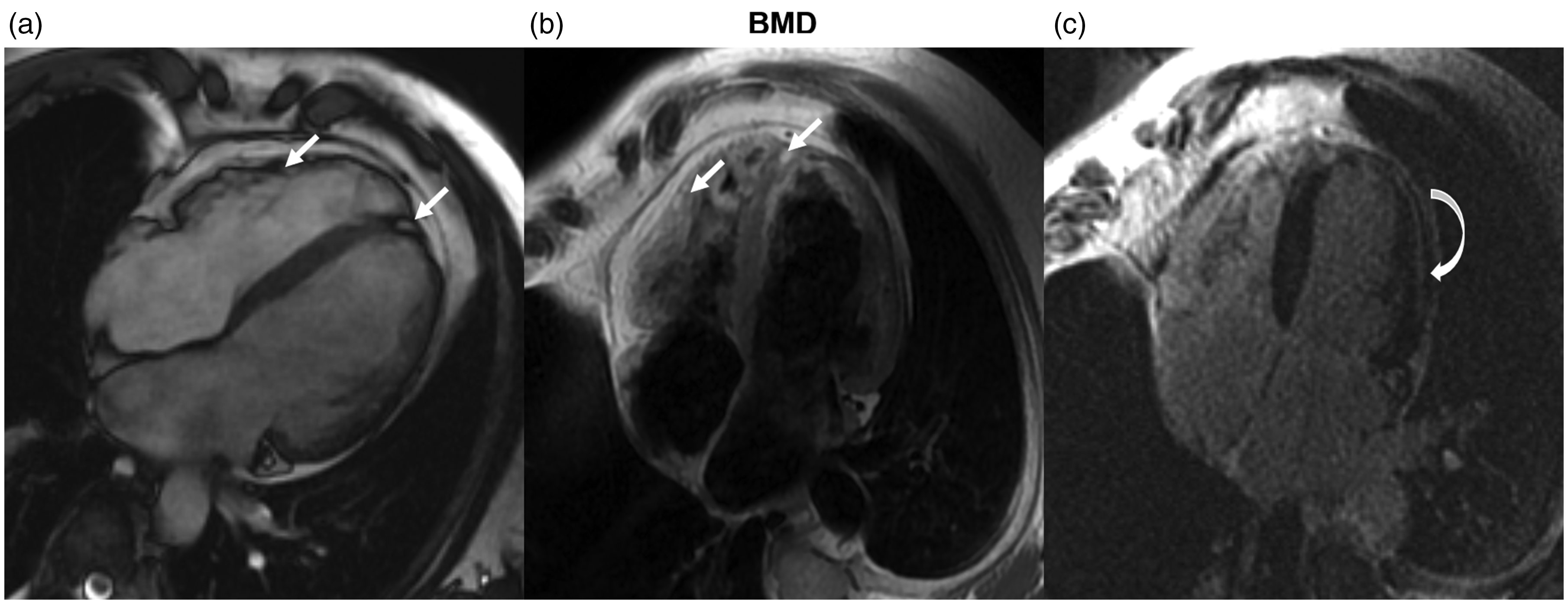
Fatty replacement in a patient with Becker muscular dystrophy (BMD).
In patients with DMD or BMD, CMR is probably less useful in those with severe impairment of the skeletal muscles and/or the heart, because of contraindications to CMR or poor tolerance of this examination, as well as the poor prognosis and the small likelihood that CMR findings will change the therapeutic strategy. Conversely, CMR is particularly promising for early diagnosis of cardiac involvement across a wide range of conditions, from known carriers of disease-causing mutations to patients undergoing a diagnostic work-up for skeletal myopathy of unknown origin. While LVEF can usually be at least estimated on echocardiogram even in patients with poor acoustic windows, CMR allows a much more accurate measurement. Patients with systolic dysfunction (LVEF <40%) can then start a therapy of neurohormonal antagonism, which might have a positive impact on the progression of cardiac disease. Furthermore, the finding of extensive LGE could warrant the start of the same therapy even when systolic function is preserved or mildly depressed, or at least suggests the need for a closer follow-up. A more sophisticated analysis of cardiac structure and function through, for example, ECV quantification or myocardial tagging (but possibly also fat–water separation imaging, blood oxygenated level dependent imaging, spin labelling or other cutting-edge techniques), might enable an earlier detection of cardiac involvement, but the clinical relevance of these findings is currently unclear.
Given the limited inter and intra-observer variability of this technique, serial CMR examinations could be considered to assess the changes in cardiac structure and function over time, and then possibly also the response to therapy. From the perspective of clinical research, follow-up studies would be important also to clarify disease evolution and the prognostic value of baseline CMR features. Along the same line of reasoning, a CMR characterisation including at least native or post-contrast T1 mapping analysis and myocardial tagging would be valuable to assess the potential of these techniques providing quantitative measures of cardiac structure and function. Future studies could also assess abnormal energy metabolism and fibro-fatty replacement in the heart (Figure 4). In parallel, technological advances reducing scan times and allowing free-breathing acquisitions will be likely to increase the feasibility of CMR examination in patients with MD.
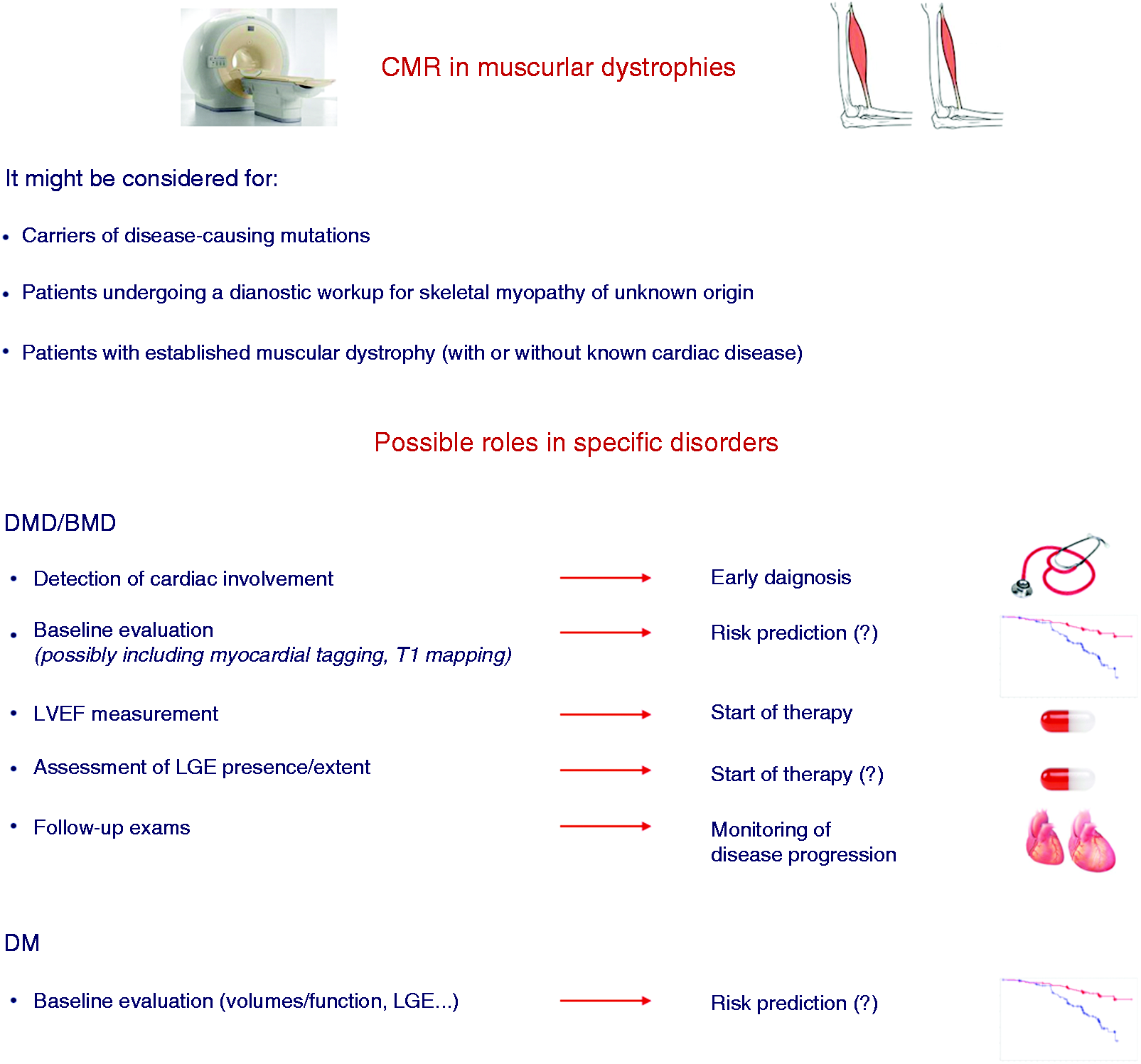
Possible roles of cardiac magnetic resonance (CMR) in patients with muscular dystrophies.
The possible role of CMR findings, particularly LGE presence and extent and fibro-fatty infiltration, for the prediction of arrhythmic complications remains to be established. Particularly for patients with a higher risk of an arrhythmic SCD (such as patients with DM, EDMD and LGMD type 1B), the challenge is to identify predictors of SCD through CMR. Similarly, searching for CMR predictors of conduction disturbances, and then the need for prophylactic pacemaker implantation, is warranted. Gathering further data on both topics, possibly in the setting of large-scale multicentre collaborations, is advisable.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.