
Abstract
During the 1990s, three Directives were enacted that aimed to harmonise the regulation of medical devices within the European Union (EU). Custom-made devices (CMDs) were subject to Council Directive 93/42/EEC of 14 June 1993 concerning medical devices (Medical Device Directive [MDD]), which was given effect in the UK by the Medical Devices Regulations 2002 (UK MDR 2002). Regulation (EU) 2017/745 (Medical Device Regulation [EU MDR]) replaced the MDD and was transposed into the Medical Devices (Amendment etc.) (EU Exit) Regulations 2019 in the UK. The UK left the EU on 31 January 2020 and entered an 11-month implementation period (IP), during which any new EU legislation that was enacted also took effect in the UK. The EU MDR was scheduled to be fully implemented on 26 May 2020 (during the IP) but this was deferred for one year, until 26 May 2021 (after the IP had concluded), as a result of the coronavirus disease 2019 (COVID-19) pandemic. Consequently, the EU MDR was removed from the UK statute book by a further amendment to the UK MDR 2002, the Medical Devices (Amendment etc.) (EU Exit) Regulations 2020. Since 1 January 2021, CMDs manufactured in Great Britain can conform to either the UK MDR 2002 (as amended) or the EU MDR (until 30 June 2023) while devices manufactured in Northern Ireland are subject to the EU MDR alone. CMDs must be supplied with a statement, a label and, depending on the risk class, instructions for use; this paper answers ten questions regarding this documentation following these legislative changes.
Keywords
Learning Objectives
To understand that custom-made devices must be supplied with the appropriate documentation
To recognise that the statement that must accompany a custom-made device must be made available to the patient
To appreciate which dental appliances are classed as custom-made devices
Introduction
During the 1990s, three Directives were enacted that aimed to harmonise the regulation of medical devices within the European Union (EU):
Council Directive 90/385/EEC of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices (Active Implantable Medical Devices Directive [AIMDD]) 1
Council Directive 93/42/EEC of 14 June 1993 concerning medical devices (Medical Device Directive [MDD]) 2
Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices (In Vitro Diagnostic Medical Devices Directive [IVDMDD]) 3
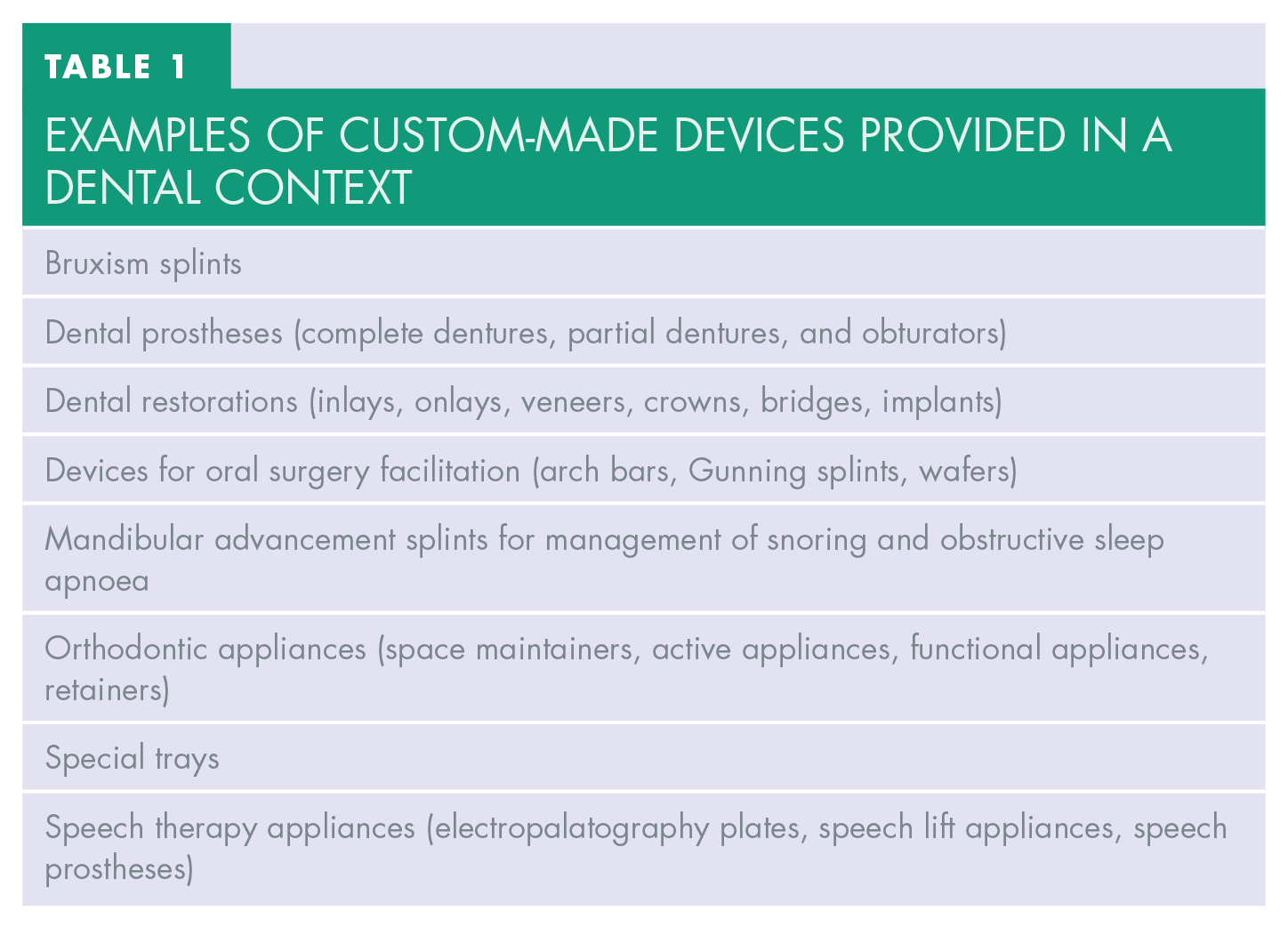
Dental professionals manufacture and provide an array of custom-made devices (CMDs) (see Table 1); these were subject to the MDD and given effect in the UK by the Medical Devices Regulations 2002 (Statutory Instrument 2002/618 [UK MDR 2002]). 4 In the years that followed, incidents with defective medical devices uncovered weaknesses in this Directive. 5 This led to the publication of Regulation (EU) 2017/745 (Medical Device Regulation [EU MDR]) 6 on 5 April 2017, which came into force on 25 May 2017 and would later supplant the MDD.
EXAMPLES OF CUSTOM-MADE DEVICES PROVIDED IN A DENTAL CONTEXT
On 23 June 2016, a non-legally binding referendum took place in the UK that asked the electorate whether the country should remain a member of, or leave, the EU; 48.11% of the votes cast were supportive of the former and 51.89% of the votes cast were in favour of the latter. The government of the time pledged to implement the outcome and as a result, in preparation for the UK’s departure, EU legislation was largely transposed into UK law: the Medical Devices (Amendment etc.) (EU Exit) Regulations 2019 (Statutory Instrument 2019/791 [UK MDR 2019]), 7 an amendment of the UK MDR 2002, 4 was derived from the EU MDR and was expected to come into effect on exit day.
The UK left the EU on 31 January 2020 and entered an 11-month implementation period (IP), during which EU law continued to apply.5,8 Full implementation of the EU MDR was originally scheduled to take place on 26 May 2020 (during the IP). It was therefore anticipated that the new Regulations would be implemented in the UK and efforts were made to prepare dental professionals for the change.9-14
However, on 23 April 2020, Regulation (EU) 2020/561 (an amendment to the EU MDR) was adopted, which deferred the full implementation of the EU MDR until 26 May 2021 so that efforts could be concentrated on the response to the coronavirus disease 2019 (COVID-19) pandemic. 15 Therefore, full implementation of the EU MDR ultimately took place after the IP had concluded and on 8 December 2020, the UK MDR 2002 was further updated by the Medical Devices (Amendment etc.) (EU Exit) Regulations 2020 (Statutory Instrument 2020/1478 [UK MDR 2020]), 16 which substituted “exit day” for “IP completion day” (which was 1 January 2021) and removed the provisions of the EU MDR, meaning that the UK MDR 2002 4 (essentially the MDD) continues to apply.
Despite the name, the UK MDR does not now apply to all constituent countries of the UK; it is only applicable in Great Britain (England, Scotland and Wales). In Northern Ireland (NI), the EU MDR has been implemented due to the Protocol on Ireland/Northern Ireland which provides NI with access to the EU’s single market and means that medical device manufacturers in NI are required to follow the EU legislation. 17
A paper published in this journal in June 2022 18 presented ten common questions regarding CMD documentation requirements following these legislative changes, together with their answers. This paper answers some additional queries on the topic.
1. Are manufacturers required to make the statement that accompanies a CMD available to the patient?
The concept of the statement originated in the MDD and specifies that:
“Class IIa, IIb and III devices shall be accompanied by the statement referred to in Annex VIII.”
— MDD Article 4(2)
2
The comparative text in the UK MDR 2002
4
states that:
“A custom-made device—
(a) in respect of which the conditions specified in Annex VIII are satisfied; and
(b) in the case of a Class IIa, Class IIb and Class III device, which is accompanied by the
statement required by Section 1 of Annex VIII,
shall be taken to comply with the relevant essential requirements unless there are reasonable grounds for suspecting that the device does not comply with those requirements.”
— UK MDR 2002 regulation 9(5)
4
On 5 September 2007, an amendment to the MDD, Directive 2007/47/EC (MDD 2007),
19
was published, which amended MDD Article 4(2) and clarified that the statement needs to be made available to the patient:
“Class IIa, IIb and III devices shall be accompanied by the statement referred to in Annex VIII, which shall be available to the particular patient identified by name, an acronym or a numerical code.”
Medical device manufacturers were required to be compliant with this amendment by 21 March 2010. MDD 2007 was implemented in UK law in the Medical Devices (Amendment) Regulations 2008 (Statutory Instrument 2008/2936 [UK MDR 2008]),
20
which amended UK MDR 2002 regulation 9(5):
“In regulation 9 of the principal Regulations (determining compliance of general medical
devices with relevant essential requirements)—
(a) after paragraph (5), insert—
‘(5A) When a custom-made device is supplied to a patient, the healthcare professional
who writes the prescription for the custom-made device shall, in relation to each patient that
they supply with such a device—
(a) ensure that the patient is aware that they may request the statement containing the
information required by Sections 1 and 2 of Annex VIII; and
(b) ensure that the statement containing the information required by Sections 1 and 2
of Annex VIII is made available to the patient on request.’”
— UK MDR 2008 regulation 6(a)
20
The UK MDR 2008 was published on 12 November 2008, laid before Parliament on 19 November 2008, and came into force on 21 March 2010.
Under the EU MDR, this requirement remains the same:
“Custom-made devices shall be accompanied by the statement referred to in Section 1 of Annex XIII, which shall be made available to the particular patient or user identified by name, an acronym or a numerical code.”
— EU MDR Article 21(2)
6
To summarise, under current UK and EU legislation, CMD manufacturers are obligated to make the statement that accompanies a CMD available to the patient.
2. Are manufacturers based in Great Britain permitted to manufacture CMDs in accordance with, and provide documentation that makes reference to, the EU MDR?
The EU MDR came into force while the UK was an EU member state. It was anticipated that the EU MDR would be fully implemented during the IP, and thereby enter UK legislation. Consequently, dental professionals in Great Britain may have begun to manufacture CMDs in accordance with the EU MDR and developed documentation (most pertinently the statement) that makes reference to the EU Regulation. Medical devices that are manufactured in Great Britain can be made in accordance with the EU MDR, and hence use documentation that makes reference to it, until 30 June 2023. 21
3. Should the documentation that accompanies a CMD carry signatures of the prescriber and/or manufacturer?
Under current UK and EU legislation, there is no requirement for the documentation that accompanies a CMD to include the signatures of the prescriber or manufacturer of the device, but it does need to include their names.
4. Can the prescriber of a CMD use their own prescription, or does it need to be issued by a dental laboratory?
Under current UK and EU legislation, a CMD must be manufactured “in accordance with a written prescription” but there are no stipulations with regards to the format with which this prescription must take so the prescriber is not obligated to use a laboratory issued prescription. In addition, guidance published by the Medicines and Healthcare products Regulatory Agency (MHRA), the executive agency of the Department of Health and Social Care that regulates medicines and medical devices, states that:
“A written prescription may take the form of a letter from a qualified person or a moulded impression of the shape of the required device together with the order specifying customer details, and a request to ‘make as pattern’.”
— Custom-made devices in Great Britain (MHRA guidance)
22
5. For what period of time should a manufacturer retain a copy of the statement that accompanies a CMD?
CMD manufacturers are required to retain a copy of the statement. Under the MDD and the UK MDR 2002 the statement retention period was a minimum of five years:
“The information contained in the declarations concerned by this Annex shall be kept for a period of time of at least five years. In the case of implantable devices the period shall be at least 15 years.”
— MDD Annex VIII(13.1)
2
“No person shall supply a custom-made device (if that supply is also a placing on the
market, or if that supply is of a custom-made device that has been placed on the market) unless its manufacturer or his authorised representative—
(a) has drawn up a statement containing the information required by Sections 1, 2 and
2.1 of Annex VIII;
(d) keeps available for the Secretary of State, for a minimum period of five years, the
information contained in the statement referred to in paragraph (a). . .”
— UK MDR 2002 regulation 15(a) and (d)
4
Under the EU MDR, the retention period for a copy of the CMD statement has changed to at least ten years:
“The statement referred to in the introductory part of Section 1 shall be kept for a period of at least 10 years after the device has been placed on the market. In the case of implantable devices, the period shall be at least 15 years. Section 8 of Annex IX shall apply.”
— EU MDR Annex XIII(4)
6
Therefore, a statement for a CMD that is manufactured in Great Britain should be kept for a period of five years but a statement for a CMD that is manufactured in the EU or NI should be retained for at least ten years.
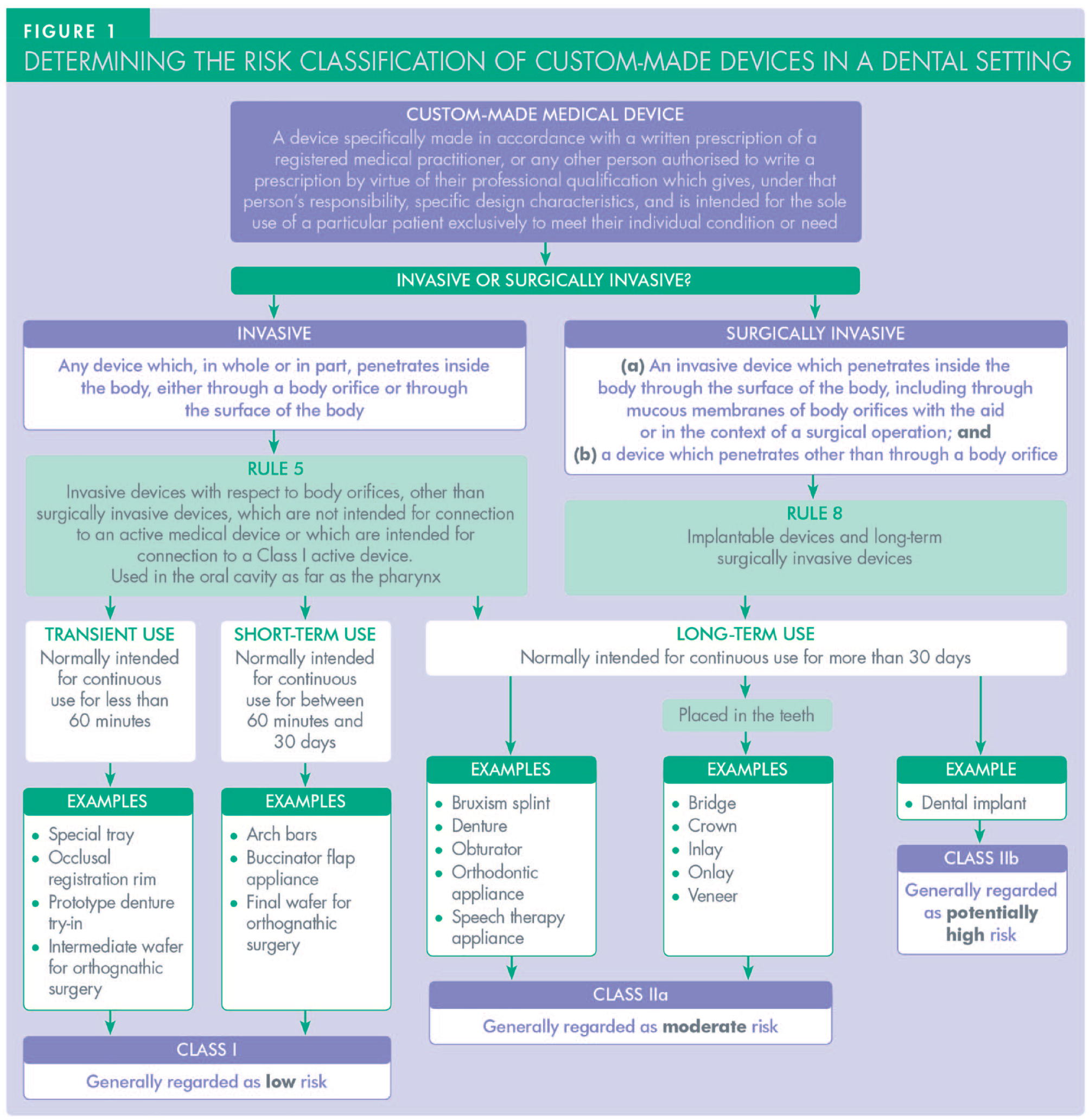
6. Should occlusal registration rims or prototype denture try-ins be provided with a CMD statement?
Under UK and EU medical device legislation, all devices are categorised according to the level of risk they are expected to present, their anticipated duration of use and whether they are invasive or surgically invasive. As Figure 1 demonstrates, CMDs such as occlusal registration rims and prototype denture try-ins, which are invasive but not intended to be used for more than 60 minutes, are categorised as Class I. The MDD states that:
“Class IIa, IIb and III devices shall be accompanied by the statement referred to in Annex VIII, which shall be available to the particular patient identified by name, an acronym or a numerical code.”
UK legislation includes the same requirement:
“in the case of a Class IIa, Class IIb and Class III device, which is accompanied by the statement required by Section 1 of Annex VIII, shall be taken to comply with the relevant essential requirements unless there are reasonable grounds for suspecting that the device does not comply with those requirements.”
— UK MDR 2002 regulation 9(5)
4
In addition, MHRA guidance dated 31 December 2020 states that:
“It should be noted that the statement does not need to be provided with a custom-made device which has been classified as class I.”
— Custom-made devices in Great Britain (MHRA guidance)
22
This means that occlusal registration rims and prototype denture try-ins that are manufactured and provided in Great Britain do not need to be accompanied by a statement. This class stipulation does not appear in the comparable sentence within the EU MDR:
“Custom-made devices shall be accompanied by the statement referred to in Section 1 of Annex XIII, which shall be made available to the particular patient or user identified by name, an acronym or a numerical code.”
— EU MDR Article 21(2)
6
Therefore, Class I CMDs that are manufactured and provided to patients in the EU and NI should be supplied with a statement. However, since occlusal registration rims and prototype denture try-ins are only in the oral cavity momentarily during the denture construction process, rather than functional devices that are provided to the patient, it is acceptable to only provide the statement with the completed denture.
7. Should sports mouth guards be provided with CMD documentation?
Mouthguards (also known as gumshields, mouth protectors or sports guards) cover the teeth and surrounding mucosa with the aim of preventing or reducing trauma to the teeth, gingival tissue, lips and jaws.
23
MHRA Guidance states that: “mouth guards are only medical devices when intended for a specific ‘medical’ purpose,
for example as a retainer following orthodontic treatment or for use in the treatment of
sleep apnoea or for bruxism. In most other cases these products will be PPE, including
those intended for sports purposes.”
— Borderlines with medical devices and other products in Great Britain (MHRA guidance)
24
Therefore, sports mouth guards are outside the jurisdiction of medical device legislation and do not need to be provided with CMD documentation. Sports mouth guards are instead subject to Personal Protective Equipment legislation. Sports mouth guards manufactured in Great Britain are governed by the Personal Protective Equipment (Enforcement) Regulations 2018 (Statutory Instrument 2018/390) 25 while those manufacturers of sports mouth guards in the EU or NI must instead follow Regulation (EU) 2016/425. 26
8. Should bleaching trays be provided with CMD documentation?
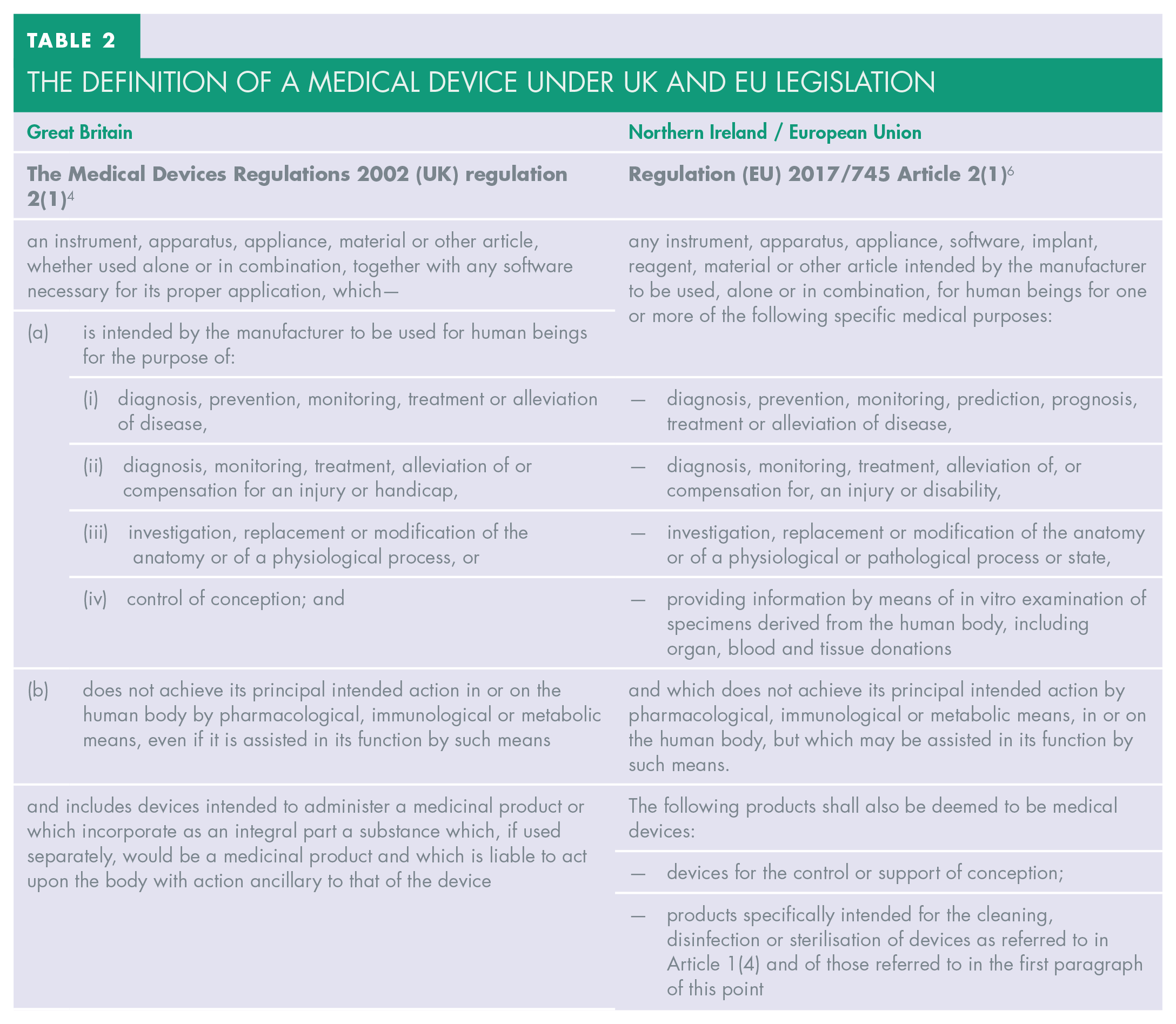
Bleaching trays, also known as tooth whitening trays, do not need to be provided with this documentation as they do not fulfil the definition of a medical device under UK and EU medical device legislation (see Table 2). Such devices are categorised as cosmetic products and were subject to Council Directive 76/768/EEC of 1976-07-27 on the approximation of the laws of the Member States relating to cosmetic products, 27 which was replaced by Regulation EC 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products. 28 Since 1 January 2021, cosmetic products in Great Britain have been governed by Schedule 34 of the Product Safety and Metrology etc. (Amendment etc.) (EU Exit) Regulations 2019 (Statutory Instrument 2019/696) 29 while those in the EU and NI remain subject to Regulation EC 1223/2009. 28
THE DEFINITION OF A MEDICAL DEVICE UNDER UK AND EU LEGISLATION
9. Should the documentation that accompanies a CMD include the manufacturer’s Competent Authority reference number?
The MDD stated that:
“Member States may require that the manufacturer shall submit to the competent authority a list of such devices which have been put into service in their territory”
— MDD Article 11(6)
2
This remains the same under the EU MDR:
“Member States may require that the manufacturer of a custom-made device submit to the competent authority a list of such devices which have been made available in their territory”
— EU MDR Article 21(2)
15
In the UK, it is a legal requirement for medical device manufacturers to register with the Competent Authority (CA):
“(1) Subject to paragraph (6), for the purpose of enabling the Secretary of State to exercise his functions under these Regulations, any person to whom this paragraph applies shall—
(a) inform the Secretary of State of the address of his registered place of business; and
(b) supply the Secretary of State with a description of each category of device concerned.
(2) Paragraph (1) applies to—
(a) a manufacturer with a registered place of business in the United Kingdom who, under his own name, places on the market in the United Kingdom a relevant device which is a Class 1 device or a custom-made device, other than a system or procedure pack which is not CE marked; and
(b) a person with a registered place of business in the United Kingdom who sterilises before use relevant devices designed by their manufacturer to be sterilised before use.”
— UK MDR 2002 regulation 19
4
The CA for the UK is the MHRA, which issues each registered manufacturer with a unique CA reference number. There is no obligation for this number to be included on the documentation that accompanies a device. However, the presence of a CA reference number on CMD documentation expedites the process of determining whether or not a manufacturer is registered with the MHRA.
10. In cases where the prescriber is an oral and maxillofacial surgeon, should the documentation that accompanies a CMD include their General Medical Council (GMC) registration number?
Where the prescriber and/or manufacturer of a CMD is a General Dental Council (GDC) registrant, the documentation that accompanies the device must include their GDC registration number(s).18,30 Oral and maxillofacial surgeons are GMC registrants, so may not be GDC registrants. 31 GMC guidance states that it is good practice for registrants to display their GMC reference number on medical records (Table 3). 32
GENERAL MEDICAL COUNCIL (GMC) GUIDANCE ON USING THE GMC REFERENCE NUMBER 30
Conclusion
This paper has provided an overview of some of the issues relating to the documentation that needs to be supplied with CMDs following a period of regulatory change. This paper should not be referred to on its own and dental professionals who manufacture CMDs should refer to the appropriate legislation.