
Abstract
Objective
This study aims to investigate the causal relationship between SLC16A1 lactylation modification and systolic blood pressure (SBP) and diastolic blood pressure (DBP), as well as the potential mediating role of lipidomics-triglyceride (50:3/50:1) in this association.
Methods
This study combines eQTL data and lactylation modifications to identify 16 associated genes, using Mendelian randomisation (MR) to assess their effects on SBP and DBP. It also employs summary-data-based Mendelian randomisation (SMR) for validation and lipidomics analysis to explore the effect of SLC16A1 lactylation on triglyceride (TG) subtypes (50:3/50:1). Finally, mediation analysis quantifies the role of triglyceride subtypes in the link between SLC16A1 lactylation and blood pressure changes.
Results
SLC16A1 lactylation modification negatively correlates with SBP (β = -1.181, P < 0.001) and DBP (β = -0.9954, P < 0.001), confirmed by SMR analysis (P < 0.001). Lipidomics shows TG(50:3) and TG(50:1) are crucial for SBP and DBP, with TG(50:3) mediating 11.32% of the effect on SBP (P = 0.0381) and TG(50:1) mediating 9.32% on DBP (P = 0.00066).
Conclusion
This study reveals that SLC16A1 lactylation modification might regulate SBP and DBP by suppressing the levels of TG(50:3) and TG(50:1), offering potential therapeutic targets for precision interventions in metabolic hypertension.
Keywords
Introduction
Hypertension (HTN), a leading risk factor for global disease burden, remains incompletely elucidated in its pathological mechanisms. 1 Although genome-wide association studies (GWAS) have identified hundreds of blood pressure-associated genetic loci, 2 significant knowledge gaps persist in understanding how these genetic variants translate into molecular mechanisms driving blood pressure phenotypes.
Despite the wide availability of antihypertensive medications, current management of hypertension remains globally insufficient.3,4 Clinically, a significant proportion of patients develop resistant hypertension, exhibit substantial variability in drug response, or continue to face high residual cardiovascular risks despite achieving target blood pressure levels.5–7 These clinical gaps highlight that classical hemodynamic perspectives do not fully capture the complexity of the disease. While traditional antihypertensive agents — such as those targeting the renin-angiotensin-aldosterone system (RAAS) or calcium channels — effectively modulate downstream hemodynamics,8–10 they often fail to correct the underlying upstream dysregulation of energy metabolism and epigenetic remodeling within vascular wall cells. This persistent metabolic and epigenetic (epimetabolic) dysregulation is increasingly recognized as a fundamental driver of drug resistance and residual cardiovascular risk.9–11 Therefore, the advances in the conceptual framework of “epimetabolic regulation” 12 provide a more profound and fundamental paradigm for dissecting the reciprocal interplay between metabolism and epigenetics and for explaining these persistent therapeutic gaps.
Lactylation, a pivotal nexus between epigenetic regulation and energy metabolism, has been demonstrated to participate in immune responses and tumor metabolic reprogramming,13–16 yet its role in cardiovascular homeostasis remains underexplored. The SLC16A1 gene encodes monocarboxylate transporter 1 (MCT1), which facilitates transmembrane transport of monocarboxylic acids such as lactate and plays central roles in energy homeostasis and tumor metabolism.17,18 Furthermore, SLC16A1 may additionally participate in diverse biological processes through epigenetic modifications including DNA methylation 19 and post-translational regulatory mechanisms such as ubiquitination. 20 Notably, a prominent lactylation modification site on the SLC16A1 protein exhibits spatial colocalization with the blood pressure-associated GWAS signal (rs1049434), 21 suggesting its potential involvement in blood pressure regulation through epimetabolic reprogramming. However, the precise molecular mechanisms and downstream effectors remain uncharacterized.
At the metabolic level, alterations in triglyceride (TG) subtype composition are closely associated with vascular dysfunction. 22 High-resolution mass spectrometry analyses reveal that structural divergences in fatty acid chain length and unsaturation between TG(50:3) and TG(50:1) 23 may confer distinct pathways for vascular tension regulation. However, critical questions remain unresolved: (1) how epimetabolic modifications selectively govern TG subtype-specific metabolic networks, and (2) how heterogeneous TG subtypes differentially modulate the dynamic equilibrium of systolic (SBP) and diastolic blood pressure (DBP). Addressing these questions may help establish TG(50:3) and TG(50:1) subtype signatures in concert with SLC16A1 lactylation status as a molecular stratification framework, which could ultimately guide more personalized antihypertensive strategies — particularly for patients with resistant hypertension or suboptimal drug response — through targeting the epimetabolic axis rather than downstream hemodynamics alone.
This study by integrating multi - omics data with Mendelian randomisation (MR) analysis investigates how SLC16A1 lactylation suppresses TG(50:3/50:1) to differentially regulate SBP/DBP, with three key contributions: (1) Proposing a “epimetabolic-blood pressure” interaction model that may contribute to the understanding of how lactylation-driven metabolic reprogramming relates to GWAS-derived blood pressure phenotypes; (2) Defining TG (50:3/50:1) subtype-specific regulatory patterns as biomarkers that may help in hypertension subclassification; (3) Suggesting SLC16A1 lactylation as a potential metabolic-vascular therapeutic target that may contribute to the understanding of unresolved mechanisms in hypertension management.
Methods
This is an analytical genetic epidemiology study employing a two-sample MR framework integrated with mediation analysis to infer causal relationships from observational data.
The study design flowchart is shown in Figure 1. This study aims to investigate the causal relationship between SLC16A1 lactylation modification and SBP and DBP, as well as the potential mediating role of lipidomics-TG(50:3/50:1) in this association. The core principle of MR is to analyze the causal relationship between exposure factors and outcome variables using instrumental variables (IV). MR analysis relies on the following three core assumptions
24
: (1) Association assumption: The instrumental variable must be significantly associated with the exposure factor; (2) Exclusion assumption: The instrumental variable must affect the outcome variable only through the exposure factor; (3) Independence assumption: The instrumental variable must be independent of the outcome variable and confounding factors (Figure 2). Schematic diagram of the research design. Created with Figdraw (https://www.figdraw.com/), copyright code: APTYP11912. Core assumptions of MR.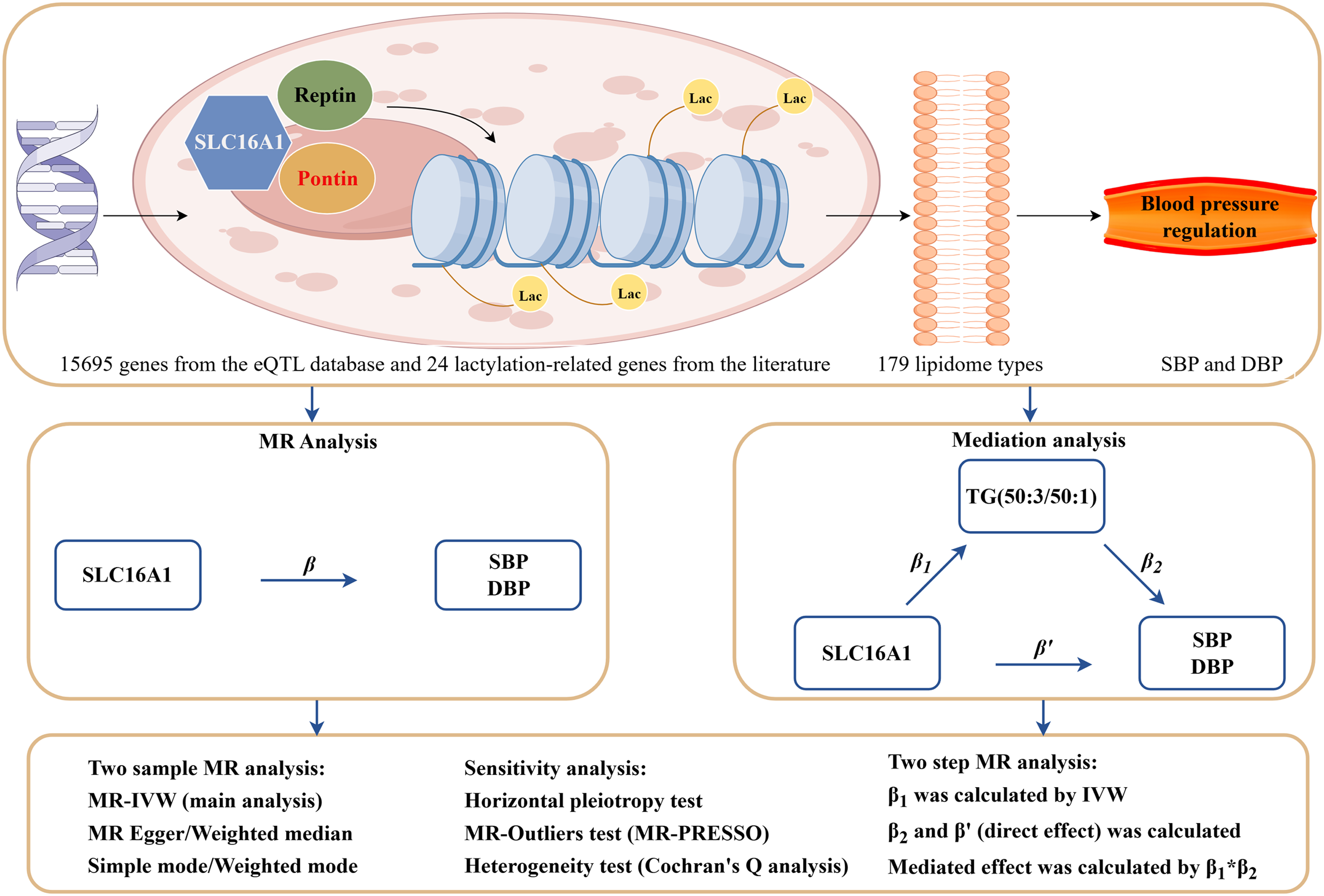

To explore the downstream mechanisms of SLC16A1 lactylation modification in the regulation of SBP and DBP, we employed the following steps: First, MR analysis was used to assess the total causal effect of SLC16A1 on SBP and DBP. Next, two-sample weighted median regression (TWMR) was applied to evaluate the direct causal effects of TG(50:3/50:1) on SBP and DBP. Following this, we assessed the causal effect of SLC16A1 on TG(50:3/50:1) (β1) and calculated the causal effect of TG(50:3/50:1) on SBP and DBP (β2). The mediation effect was then calculated, representing the influence of SLC16A1 on SBP and DBP mediated by TG(50:3/50:1). This was done using the product rule, i.e., β 1 × β 2 . Finally, we quantified the proportion of the mediation effect in the total effect of SLC16A1 on SBP and DBP by calculating the ratio of the indirect effect to the total effect.
Data selection
The eQTLGen Consortium database serves as a critical resource for elucidating the interplay between genetic regulation and complex disease pathogenesis, with substantial implications for precision medicine and genomic discovery. This comprehensive repository systematically maps genetic variants influencing phenotypic traits through downstream effects on transcriptional regulation. Utilizing blood-derived samples from 31,684 individuals, the project establishes genome-wide associations between single nucleotide polymorphisms (SNPs) and gene expression patterns. Integrated analysis of 37 independent datasets examines 11 million common SNPs and 10,317 trait-associated variants, assessing their regulatory impact on 19,960 protein-coding genes. 25
Through systematic literature review, our team identified three seminal studies elucidating lactylation-mediated epigenetic regulation. The first investigation (PMID: 39662177) characterized temporal dynamics of histone lactylation, demonstrating its pivotal role in macrophage polarization and suggesting therapeutic potential in inflammatory disorders and oncogenesis. 26 The second study (PMID: 39609834) established lactate as a central modulator of histone modification landscapes, revealing lactylation-dependent transcriptional regulation of immune responses and neoplastic proliferation. 27 The third pivotal work (PMID: 39278916) deciphered molecular mechanisms underlying lactate metabolism and histone lactylation, identifying critical functions in macrophage activation pathways and tumor immunobiology. 28 Collectively, these findings position lactylation as a metabolic sensor coordinating chromatin remodeling and gene expression programs, with profound implications for inflammation, malignancy, and immune homeostasis. To ensure methodological rigor, our inclusion criteria for lactylation-related genes were strictly literature-driven and evidence-based. Specifically, we traced every mentioned gene back to its original experimental research article, requiring definitive experimental evidence confirming its lactylation modification. This rigorous manual mining ultimately yielded 24 high-confidence lactylation-related genes.
To ensure that the genetic IV used in our subsequent MR analysis had sufficient statistical power, we employed a stringent “intersection mapping” strategy. We extracted comprehensive eQTL data encompassing 15,695 genes from the eQTLGen database, applying stringent linkage disequilibrium (LD) filtering (P < 5 × 10-8, window size = 10,000 kb, R2 = 0.1). Subsequently, we intersected the 24 manually screened lactylation-related genes with the strictly quality-controlled eQTL genes. Only genes that met a dual criterion — possessing concrete experimental evidence of lactylation and being regulated by highly significant, independent eQTLs — were retained for the downstream analysis. Given the lack of a well-established genome-wide lactylation reference database, experimentally verified evidence from published peer-reviewed studies was used as the primary benchmark for biological validation of the selected gene set. This intersection strategy maximizes both the biological validity and statistical robustness of our MR analytical framework.
For lipidomic profiling, we incorporated data from a Nature Communications GWAS (n=7,174 Finnish participants) analyzing 179 lipid species against 849,000 high-quality genetic variants. 29 This resource enables precise mapping of lipidomic determinants in cardiometabolic pathophysiology.
Blood pressure phenotypes SBP/DBP were obtained from the International Consortium of Blood Pressure (ICBP) datasets ieu-b-38 and ieu-b-39 (https://gwas.mrcieu.ac.uk/). This multi-phase GWAS meta-analysis encompassed 757,601 European participants from the UK Biobank and ICBP cohorts, providing robust genetic associations for hemodynamic traits. 30
This MR study used publicly available GWAS summary data. As the original studies were ethically approved, this analysis of anonymous data was exempt from further ethical review and informed consent. All procedures followed the Declaration of Helsinki.
Selection of instrumental variables
To satisfy the Mendelian randomization assumptions of strong instrument-exposure associations and independence from confounding factors, we implemented a rigorous multi-step genetic variant selection protocol. Genome-wide significant SNPs associated with the exposure (P < 5×10-8) were initially prioritized to ensure robust genetic instruments.31,32 Linkage disequilibrium (LD) pruning was subsequently performed to exclude SNPs within 10,000 kb regions exhibiting LD (R2 > 0.1) with the index variant, thereby mitigating bias from correlated genetic effects.
33
The proportion of exposure variance explained (R2) was calculated using the formula:
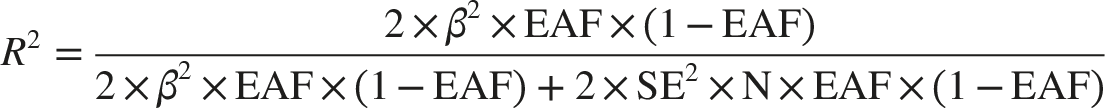
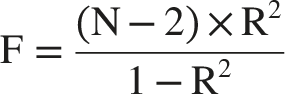
Harmonization of exposure and outcome datasets aligned effect alleles, with exclusion of palindromic SNPs (AT/TA or CG/GC polymorphisms) to prevent strand ambiguity. The LDlink platform (https://ldlink.nih.gov/?tab=ldtrait) facilitated pleiotropy screening by systematically removing variants associated with potential confounders or secondary phenotypes. 34 Final IV met stringent criteria: (1) genome-wide significance for exposure association (P < 5×10-8), (2) LD independence (R2 < 0.1), and (3) absence of pleiotropic confounding. Directional interpretation of genetic effects was based on β coefficients, where positive values (β > 0) indicated risk-increasing alleles and negative values (β < 0) denoted protective effects.
Statistical methods
MR analysis
MR analyses were conducted using the Two Sample MR package (v0.5.6) in R. Genome-wide significant single-nucleotide polymorphisms (SNPs; P < 5×10-8) were selected as IV. For single-SNP IVs, causal estimates were derived using the Wald ratio estimator. Multi-SNP analyses employed five complementary approaches: inverse variance weighted (IVW) meta-analysis, MR-Egger regression, weighted median, simple mode, and weighted mode estimators. The IVW method served as the primary analytical framework due to its conservative bias profile and statistical efficiency, while supplementary methods provided pleiotropy-robust sensitivity assessments.
Sensitivity analysis
The robustness of MR findings was systematically evaluated through sensitivity analyses. Cochran’s Q statistic was used to assess IV heterogeneity, with P < 0.05 indicating significant heterogeneity requiring random-effects models. Horizontal pleiotropy was examined via MR-Egger intercept testing (P < 0.05 threshold for pleiotropic bias). Leave-one-out analyses were performed to identify disproportionate influences of individual SNPs through sequential IV exclusion.
Summary-data-based Mendelian randomization (SMR) analysis
SMR analyses were implemented to investigate transcriptional mechanisms underlying blood pressure regulation. The Wald ratio framework was applied using single-SNP IVs from eQTL data, supplemented by heterogeneity in dependent instruments (HEIDI) testing to exclude linkage disequilibrium-confounded associations (HEIDI P > 0.05 required for causal inference).
Mediation analysis
To elucidate mediation pathways linking SLC16A1 lactylation modifications to blood pressure regulation via TG subspecies, a two-stage MR framework was employed. First, MR quantified the total effect of SLC16A1 lactylation on SBP/DBP. Subsequently, mediation decomposition involved: (1) Stage 1 MR assessing lactylation effects on TG(50:3) and TG(50:1) levels (β1); (2) Stage 2 MR estimating TG subspecies effects on SBP/DBP (β2). The indirect effect magnitude was calculated as the product term β1 × β2, with mediation proportion expressed as (indirect effect/total effect) × 100. Statistical significance of mediation pathways was evaluated through non-parametric bootstrapping (1,000 replicates; P < 0.05 threshold).
Results
To identify candidate genes associated with lactylation modification, this study first extracted eQTL data for 15,695 genes from the eQTLGen database and conducted stringent LD filtering (threshold: P < 5 × 10-8, window size = 10,000 kb, R2 = 0.1) (Supplementary Data 1). Simultaneously, a systematic literature review identified 24 key genes known to participate in lactylation modification (Supplementary Data 2).
Through Venn analysis, we identified 16 overlapping genes between the eQTL dataset and the lactylation modification gene set. These genes include SLC16A1, AARS2, EP300, HDAC1, HDAC3, KAT5, LDHA, LDHB, LDHC, SIRT1, SIRT2, SIRT3, SIRT6, SLC16A4, SLC16A7, and SMARCA4. Among them, SLC16A1 plays a crucial role in lactate metabolism and epigenetic regulation and was selected as the core target for subsequent investigations.
The results indicate a significant negative correlation between SLC16A1 lactylation modification and SBP. The IVW analysis yielded a beta value of -1.1806, with P < 0.001. Similar results were obtained in the weighted median and weighted mode analyses, and MR-Egger regression showed no evidence of horizontal pleiotropy (intercept P = 0.7721). Additionally, Cochrane’s Q test detected no significant heterogeneity (P = 0.0947) (Supplementary Material 3). These findings support the existence of a significant causal relationship between SLC16A1 lactylation modification and SBP (Figure 3).
Similarly, a significant negative correlation was observed between SLC16A1 lactylation modification and DBP. IVW analysis produced a beta value of -0.9954, with P < 0.001. The MR-Egger regression intercept P-value was 0.9268, and Cochrane’s Q test P-value was 0.0328. The weighted median, simple mode, and weighted mode analyses also produced results consistent with the IVW direction (Supplementary Material 4). These results confirm a significant causal relationship between SLC16A1 lactylation modification and DBP (Figure 3). Causal relationship analysis between SLC16A1 lactylation modification and SBP/DBP.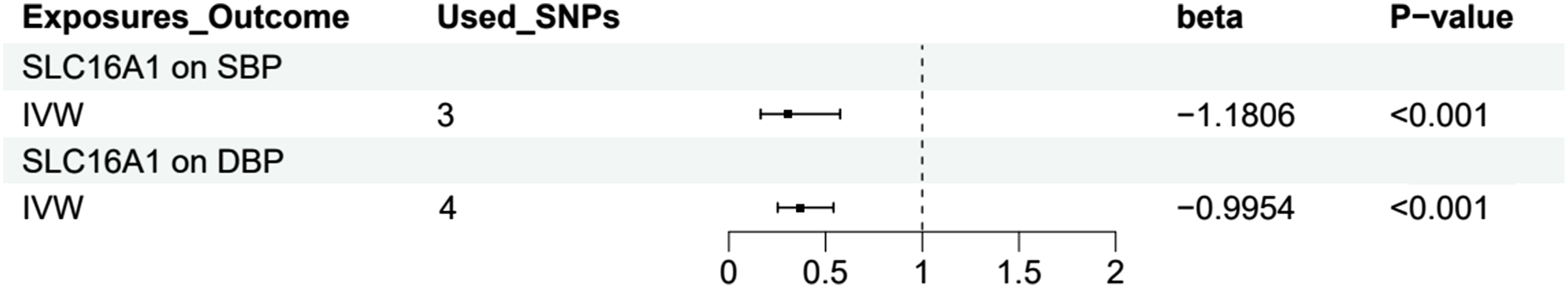
SMR analysis results of SLC16A1 and SBP.

SMR analysis results of SLC16A1 and DBP.

As shown in Table 1, rs7169 (Chr1) was identified as a significant instrumental variable for SLC16A1, with an SMR effect estimate (b_SMR) of -1.4004 (SE = 0.2601, P = 7.32 × 10-8). This indicates that for each unit increase in SLC16A1 lactylation modification, SBP decreases by 1.4004 units. The HEIDI test detected no heterogeneity (P = 0.721, nsnp_HEIDI = 20), ruling out false associations driven by genetic co-localization and supporting the robustness of the causal effect. This result is highly consistent with the previous MR analysis (IVW beta = -1.1806, P < 0.001).
For DBP (Table 2), rs7169 (Chr1) was also identified as a significant instrumental variable for SLC16A1, with an SMR effect estimate (b_SMR) of -1.1261 (SE = 0.15726, P = 8.038 × 10-13). This indicates that for each unit increase in SLC16A1 lactylation modification, DBP decreases by 1.1261 units. The HEIDI test detected no heterogeneity (P = 0.2170, nsnp_HEIDI = 20), ruling out false associations driven by genetic co-localization and supporting the robustness of the causal effect. This result is also highly consistent with the previous MR analysis (IVW beta = -0.9954, P < 0.001).
Our Mendelian randomisation analyses demonstrated significant positive causal relationships between triglyceride subspecies TG(50:3)/TG(50:1) and blood pressure phenotypes. For SBP, the IVW method revealed a positive causal effect (β = 0.4097, P = 0.0018), consistent with supplementary analyses using MR Egger (β = 0.7973, P = 0.0448), weighted median (β = 0.5722, P < 0.001), and weighted mode (β = 0.7147, P < 0.001) estimators (Figure 4). Sensitivity analyses confirmed robustness: MR-Egger regression showed no evidence of horizontal pleiotropy (intercept P = 0.2797) (Supplementary Material 5).
For DBP, outlier SNPs (rs1399627 and rs55857911) and the confounder-associated SNP rs1260326 were sequentially excluded. The IVW analysis of the final instrument set demonstrated a stronger causal association (β = 0.2725, P < 0.001), corroborated by weighted median estimates (β = 0.2006, P = 0.0385). No horizontal pleiotropy was detected (MR-Egger intercept P = 0.9), with Cochran’s Q test confirming homogeneity across variants (Supplementary Material 5). It illustrates the concordant directionality and magnitude of these causal effects across analytical methods (Figure 4). The causal associations of TG (50:3) and TG (50:1) with SBP and DBP respectively.
The results indicate a significant negative correlation between SLC16A1 lactylation modification and TG(50:3) and TG(50:1) levels (Figure 5). The effect of SLC16A1 lactylation modification on the levels of triglyceride subtypes TG (50:3) and TG (50:1).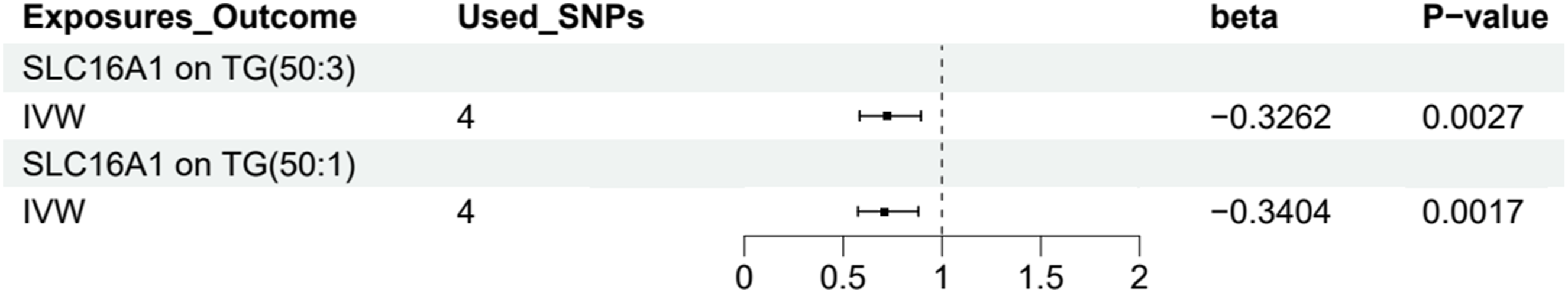
Specifically, for TG(50:3), IVW analysis yielded a beta value of -0.3262, with P = 0.0027. Similar results were obtained in the weighted median, simple mode, and weighted mode analyses, and MR-Egger regression intercept analysis showed no evidence of horizontal pleiotropy (intercept P = 0.9128). Additionally, Cochrane’s Q test detected no significant heterogeneity (P = 0.9951) (Supplementary Material 6).
For TG(50:1), IVW analysis produced a beta value of -0.3404, with P = 0.0017. The weighted median analysis also yielded similar results, and MR-Egger regression intercept analysis detected no horizontal pleiotropy (intercept P = 0.8199). Furthermore, Cochrane’s Q test detected no significant heterogeneity (P = 0.7962) (Supplementary Material 6).
Mediation effect analysis of SLC16A1 lactylation modification on SBP through TG (50:3).
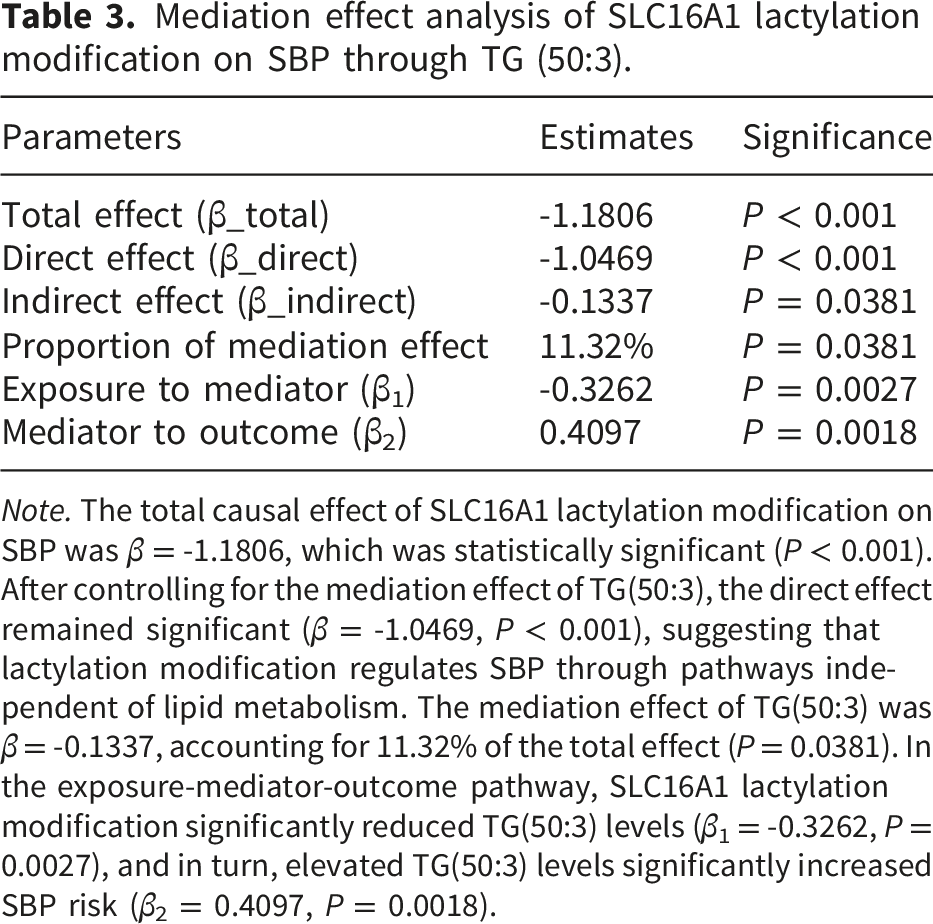
Note. The total causal effect of SLC16A1 lactylation modification on SBP was β = -1.1806, which was statistically significant (P < 0.001). After controlling for the mediation effect of TG(50:3), the direct effect remained significant (β = -1.0469, P < 0.001), suggesting that lactylation modification regulates SBP through pathways independent of lipid metabolism. The mediation effect of TG(50:3) was β = -0.1337, accounting for 11.32% of the total effect (P = 0.0381). In the exposure-mediator-outcome pathway, SLC16A1 lactylation modification significantly reduced TG(50:3) levels (β1 = -0.3262, P = 0.0027), and in turn, elevated TG(50:3) levels significantly increased SBP risk (β2 = 0.4097, P = 0.0018).
Mediation analysis identified TG(50:3) as an intermediary linking SLC16A1 lactylation modification to SBP regulation (Table 3):
SLC16A1 lactylation modification significantly reduced TG(50:3) levels (β 1 = -0.3262, P = 0.0027).
Elevated TG(50:3) levels significantly increased SBP risk (β 2 = 0.4097, P = 0.0018).
The mediation effect was estimated as β 1 ·β 2 = -0.1332 (P = 0.0381), with the mediation effect accounting for 11.32% of the total effect.
Mediation effect analysis of SLC16A1 lactylation modification on DBP through TG (50:1).
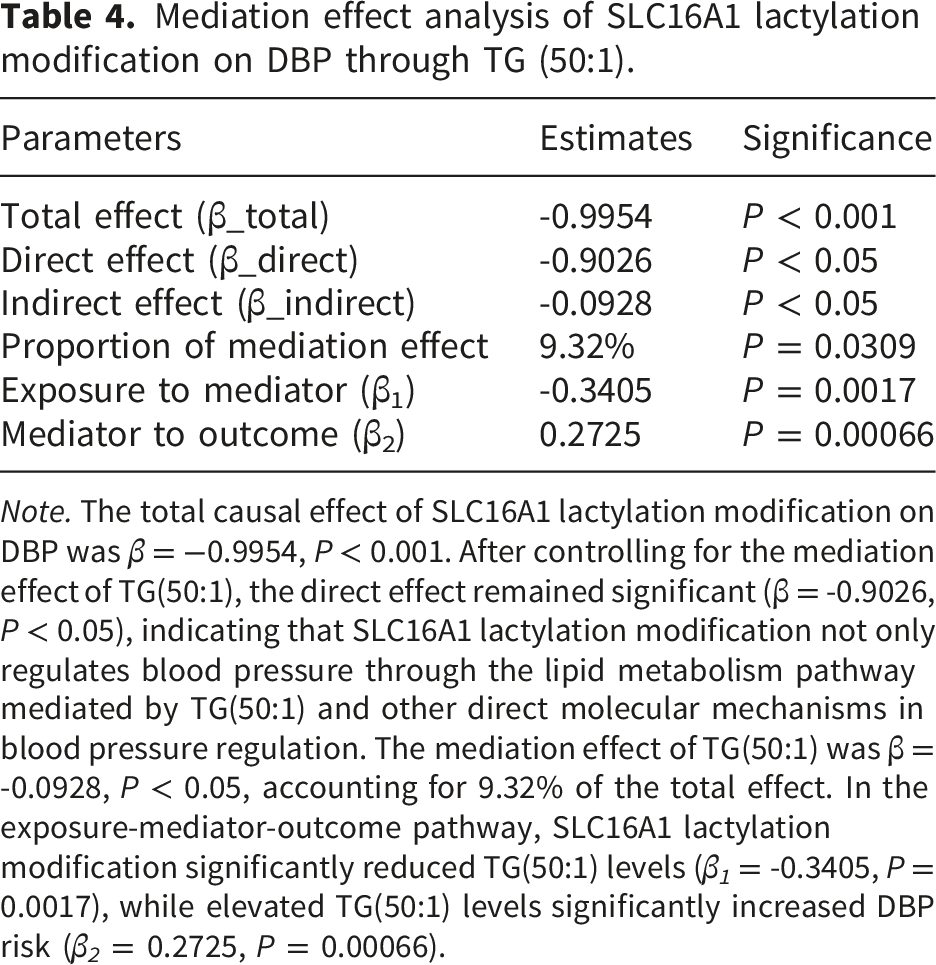
Note. The total causal effect of SLC16A1 lactylation modification on DBP was β = −0.9954, P < 0.001. After controlling for the mediation effect of TG(50:1), the direct effect remained significant (β = -0.9026, P < 0.05), indicating that SLC16A1 lactylation modification not only regulates blood pressure through the lipid metabolism pathway mediated by TG(50:1) and other direct molecular mechanisms in blood pressure regulation. The mediation effect of TG(50:1) was β = -0.0928, P < 0.05, accounting for 9.32% of the total effect. In the exposure-mediator-outcome pathway, SLC16A1 lactylation modification significantly reduced TG(50:1) levels (β 1 = -0.3405, P = 0.0017), while elevated TG(50:1) levels significantly increased DBP risk (β 2 = 0.2725, P = 0.00066).
Mediation effect analysis further identified TG(50:1) as an intermediary linking SLC16A1 lactylation modification to DBP regulation (Table 4):
SLC16A1 lactylation modification significantly reduced TG(50:1) levels (β 1 = -0.3405, P = 0.0017).
Elevated TG(50:1) levels significantly increased DBP risk (β 2 = 0.2725, P = 0.00066).
The mediation effect was estimated as β 1 ·β 2 = −0.0932 (P = 0.0309), with the mediation effect accounting for 9.32% of the total effect.
Discussion
This study integrates genetic and epimetabolic strategies to reveal that SLC16A1 lactylation modification significantly influences SBP and DBP by differentially regulating TG subtypes (such as TG(50:3) and TG(50:1)). By integrating eQTL data and lactylation modification datasets, we identified blood pressure-regulating genes and systematically explored the regulatory mechanism of SLC16A1 lactylation modification on blood pressure through MR and SMR analyses. The results demonstrated that TG(50:3) and TG(50:1) play important mediating roles in linking SLC16A1 lactylation modification to blood pressure regulation, accounting for 11.32% and 9.32% of the total effects on SBP and DBP, respectively. These findings directly address the clinical gaps in current hypertension management, particularly concerning resistant hypertension and residual cardiovascular risks. Traditional antihypertensive agents primarily target downstream hemodynamic pathways, such as the RAAS and calcium channels, 8 but accumulating evidence suggests that they often fail to reverse upstream metabolic-epigenetic dysregulation, as epigenetic modifications in hypertension do not appear to be reversible upon intensive treatment.9,10 By linking specific TG subtypes to blood pressure control, our results suggest that heterogeneous lipid profiling could aid in identifying specific metabolic-driven hypertension subphenotypes, offering new therapeutic avenues for patients misaligned with conventional care.
As a MCT1, SLC16A1 has been primarily studied in the context of tumor metabolic reprogramming (e.g., Warburg effect),35,36 whereas its role in cardiovascular diseases has been largely overlooked. Our investigation identified significant associations between SLC16A1 genetic variants and both SBP and DBP. We hypothesize that these phenotypic correlations may arise from shared causal mechanisms mediated by specific single-nucleotide polymorphisms (SNPs: rs10494151, rs7169, and rs74862275). This observation aligns with emerging paradigms in metabolic epigenetics, which reconceptualize lactate not merely as a catabolic byproduct but as a dynamic signaling modulator with systemic regulatory functions.13,37 Specifically, SLC16A1 lactylation modification may influence blood pressure through two key mechanisms. First, lipid metabolic reprogramming: As specific lipid molecules, TG(50:3) and TG(50:1) possess unique chain lengths and degrees of unsaturation, which may affect cell membrane fluidity and signal transduction. 38 Mediation analysis in this study revealed that these lipids mediate more than 11% of the effect of SLC16A1 on blood pressure, suggesting their potential involvement in vascular endothelial calcium signaling or mitochondrial function in smooth muscle cells. 39 Second, epigenetics-metabolism interplay: Lactylation modification dynamically regulates gene transcription by directly modifying histones or metabolic enzymes (e.g., KAT5). 40 This study further demonstrated that SLC16A1 lactylation modification leads to a decrease in TG(50:3) and TG(50:1) levels, likely through inhibition of lipid synthesis enzymes (e.g., DGAT1) or activation of lipolytic enzymes (e.g., ATGL). This mechanism sharply contrasts with the classical theory of lipid accumulation in obesity-associated HTN, 41 suggesting that lactylation modification may regulate vascular function through “lipid quality control” rather than “lipid quantity accumulation”.
Furthermore, recent clinical studies evaluating the relationship between lipid-related indices and cardiometabolic risk parameters provide strong supportive clinical evidence for our proposed mechanisms. For example, Bulu and Timurkaan demonstrated a significant association between atherogenic lipid indices and systemic metabolic risk parameters in diabetes, highlighting the utility of lipid-derived metrics in early stratifying cardiovascular risk. 42 Similarly, advanced lipidomics profiling has consistently proven effective in predicting cardiovascular events.43,44 Embedding our genetic-metabolic findings within such clinical observations reinforces the concept that nuanced lipid composition, such as atherogenic index variations, intimately dictates cardiovascular homeostasis and systemic vulnerability in metabolic disorders.45,46
Compared with previous studies, this study makes a significant advancement by integrating the spatiotemporal specificity of lactylation modification with the structural specificity of lipid molecules, unveiling a fine-tuned metabolic regulation network for blood pressure control. While prior studies have reported an association between lactate levels and vascular endothelial dysfunction, 47 how lactylation modification differentially regulates blood pressure via specific lipid molecules (e.g., distinct effects on SBP vs. DBP) remains unknown. By employing mediation effect models and MR causal inference, this study fills this gap and provides a molecular explanation for the heterogeneity of HTN.
Despite its novelty, this study has several limitations that warrant caution. First, population and environmental constraints: The data were primarily derived from European populations, and differences in linkage disequilibrium patterns across ethnic groups may introduce MR biases. 48 Second, mechanistic limitations: Although statistical analyses support the mediating role of TG(50:3) and TG(50:1), the specific biological pathways (e.g., involvement of lipid rafts signaling or mitochondrial phospholipid remodeling) require further experimental validation. Additionally, we fully acknowledge the inherent limitations of our annotation-based and manual literature-mining approach for identifying lactylation targets. Due to the current lack of a comprehensive genome-wide lactylome database, this strategy is inevitably subject to publication bias; genes extensively explored in popular research fields are more likely to be annotated, which may cause potential novel lactylation targets in cardiovascular physiology to be inadvertently overlooked. Furthermore, manual literature mining cannot capture the complete landscape of the lactylome, potentially underestimating the true scale of biological regulatory networks. Finally, lack of dynamic regulation analysis: This study was based on cross-sectional data, which cannot capture the temporal dynamics of lactylation modification and blood pressure regulation. Future studies incorporating longitudinal cohorts or interventional trials may help elucidate these dynamic regulatory mechanisms.
Conclusion
SLC16A1 lactylation modification differentially influences SBP and DBP by regulating TG(50:3) and TG(50:1) levels, providing a novel molecular target for precision intervention in metabolic HTN. Our findings offer a new perspective on the pathogenesis of HTN and lay the groundwork for the development of personalized treatment strategies in the future.
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Supplemental material
Supplemental material - SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation
Supplemental material for SLC16A1 lactylation links epimetabolic reprogramming of triglycerides (50:3 and 50:1) to blood pressure regulation by Weikun Zhao, Xiaoping Li, Rongjie Huang, Zuchun Luo in Sage Open Medicine
Footnotes
Acknowledgements
The authors wish to acknowledge the work of the First Affiliated Hospital of Guangxi Medical University. We extend our sincere gratitude to Professor Feng Huang from The First Affiliated Hospital of Guangxi Medical University for his expert assistance in English language editing and manuscript polishing. We also thank the platform Figdraw for their assistance in creating the schematic diagrams. Finally, we thank the Editor and the anonymous reviewers for their highly insightful and constructive feedback, which significantly elevated the quality of this manuscript.
Authors contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the Guangxi Natural Science Foundation (2025GXNSFHA069077), the Self-funded Research Project of Guangxi Zhuang Autonomous Region Administration of Traditional Chinese Medicine (GXZYC20240258 and GXZYC20240401), the Science Research and Technology Development Project of Qingxiu District of Nanning (2018038), the Key Research and Development Program of Guangxi Zhuang Autonomous Region (Guike AB24010139) and the National Natural Science Foundation of China (82360092).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Trans-eQTL data were obtained from the eQTLGen Consortium (https://www.eqtlgen.org; 31,684 individuals, 37 cohorts) analyzing 11 million SNPs and 19,960 protein-coding genes. Lipidomic profiling data originated from the Finnish genome-wide plasma lipidome study (n=7,174; https://pubmed.ncbi.nlm.nih.gov/37907536/) identifying 495 genetic associations across 179 lipid species. Blood pressure traits (SBP/DBP) were sourced from the ICBP GWAS meta-analysis (![]() ; 757,601 Europeans).
; 757,601 Europeans).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.