
Abstract
Objective
Severe acute pancreatitis (SAP) is a common emergency condition associated with high mortality. Intestinal barrier dysfunction plays a critical role in the pathogenesis of SAP. Mammalian sterile 20-like kinase 1 (Mst1) has been shown to coordinately regulate autophagy and apoptosis in cardiac and aging-related diseases. However, the precise role of Mst1 in SAP-induced intestinal barrier dysfunction remains largely unknown. This study aimed to investigate the pathophysiological impact of Mst1 on SAP-induced intestinal barrier dysfunction.
Methods
Mst1-knockout and wild-type mice were challenged intraperitoneally with caerulein combined with lipopolysaccharide (LPS) to establish an experimental SAP model. TNF-α-stimulated MODE-K cells were used to analyze the impact on autophagy and apoptosis and to elucidate the underlying mechanisms.
Results
Mst1 knockout up-regulated tight junction proteins, alleviated apoptosis and enhanced autophagy in the ileocolic mucosa tissue of SAP mice, which consequently improved cumulative survival and alleviated intestinal barrier dysfunction. Conversely, Mst1 overexpression inhibited autophagy and promoted apoptosis in TNF-α-stimulated MODE-K cells.
Conclusion
Mst1 plays an important role in SAP-related intestinal barrier dysfunction by inhibiting autophagy and enhancing apoptosis.
Introduction
Severe acute pancreatitis (SAP) is a common emergency severe syndrome with a high risk of triggering systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS), which greatly increase the mortality of patients.1,2 Despite the complex pathological mechanisms, the integrity of the intestinal mucosal barrier directly affects the progression and prognosis of SAP. 3 Apoptosis of intestinal mucosal epithelial cells disrupts structural integrity and enhances mucosa permeability, thereby severely compromising intestinal barrier function. Consequently, the translocation of intestinal bacterial and endotoxins leads to excessive neutrophils activation and secondary infections in the pancreas and other organs, ultimately exacerbating SIRS and MODS. 4 Therefore, elucidating the molecular mechanisms underlying intestinal mucosal barrier dysfunction during SAP may provide novel targets for prevention and therapy.
Emerging data indicates that autophagy regulates the function of intestinal mucosal barrier via inhibiting apoptosis, attenuating oxidative stress and improving the permeability of intestinal epithelial.5,6 Enhancing autophagy alleviates intestinal mucosal barrier dysfunction and reduces the severity of SAP.7,8 Mammalian sterile 20-like kinase 1 (Mst1), an important component of Hippo signaling pathway, has been proven to coordinately regulate autophagy and apoptosis in cardiac and aging-related diseases.9–11 Mst1 inhibits autophagy and promotes apoptosis in cardiac microvascular endothelial cells, thereby participating in the pathogenesis of diabetic coronary microvascular dysfunction. 12 Furthermore, Mst1 knockout alleviates angiotensin II-induced cardiac injury by enhancing cardiomyocyte autophagy. 13 However, the role of Mst1 in intestinal mucosal cells during SAP remains unclear. Given the importance of intestinal epithelial integrity for maintaining intestinal barrier function, it is imperative to investigate the function and mechanism of Mst1 in SAP-induced intestinal barrier injury. In the present study, we investigated the role of Mst1 in SAP-induced intestinal barrier dysfunction in vivo and in tumor necrosis factor-α (TNF-α)-stimulated intestinal mucosal cell injury in vitro. Our results show that Mst1 inhibits autophagy and promotes apoptosis in intestinal mucosal cells, thereby contributing to the pathogenesis of SAP-induced intestinal barrier dysfunction.
Methods
Materials and animals
Mst1 knockout (Mst1−/−, C57BL/6 background, generation N10) mice and their wild-type littermates (WT) were obtained from K&D gene technology (WuHan, China) and genotyped by Western blotting and real-time PCR. All mice were housed in a conventional facility with 12h dark-light cycles and allowed free access to adequate food and water. All animal experiments were strictly performed in accordance with the guidelines for the Care and Use of Laboratory Animals formulated by the Laboratory Animal Welfare and Ethics Committee of Northwest University (Xi’an), as well as the National Research Council’s Guide for the Care and Use of Laboratory Animals, and the ARRIVE guidelines. This study was formally approved by the Laboratory Animal Welfare and Ethics Committee of Northwest University (Xi’an) (Approval No.: NWU-LAEC-XA-2023-071). All procedures were designed to minimize animal suffering and discomfort, and every effort was made to reduce the number of experimental animals used, in strict adherence to the 3R principle.
Caerulein (#CM05482) were purchased from Proteintech Group, Inc. (Wuhan, China). LPS (#L4524), bafilomycin A1 (#88899-55-2) and TNF-α (Escherichia coli lipopolysaccharide, 055: B5, #L2880) were purchased from Sigma-Aldrich Inc. (St. Louis, MO, USA). α-Amylase assay kit (#C016-1-1), lipase assay kit (#A054-1-1), endotoxin assay kit (#E039-1-1), diamine oxidase (DAO) assay kit (#A088-1-1) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Mouse TNF-α ELISA kit (#CSB-E04741m), interleukin 1β (IL-1β) ELISA Kit (#CSB-E08054m), and interleukin 6 (IL-6) ELISA Kit (#CSB-E04639m) were obtained from CUSABIO Biology, Inc. (Wuhan, China). TUNEL detection kit (#C1088) were obtained from Beyotime Biotechnology Inc. (Shanghai, China). Anti-cleaved caspase-3 (#ab214430), anti-bcl-2 (#ab16904), anti-bax (#ab182734), anti-Mst1 (#ab51134), anti-beclin-1 (#ab207612), anti-p62 (#ab240635), anti-ZO-1 (#ab307799), anti-Claudin 1 (#ab307692), and anti-Occludin (#ab216327) antibodies were obtained from Abcam (Cambridge, UK). Anti-p-Mst1 (#49332) and LC3A/B (#4108) were purchased from Cell signaling (Danvers, MA, USA).
Experimental SAP animal model and survival study
The SAP mouse model was established as previously reported.14–16 Briefly, mice received intraperitoneal injections of caerulein (50 μg/kg per hour) for 10 times, with LPS (10 mg/kg) injected intraperitoneally at the time of the last caerulein injection. Control mice received equivalent volumes of saline. SAP mice were sacrificed by intracardiac puncture to collect blood samples for biochemical and pathological assessments at 48 hours after the first caerulein injection. For survival study, the living condition of SAP mice was closely monitored, and the mortality was recorded twice daily for 5 days after the first caerulein injection. Littermate mice were used in the survival study. Each group contained 15 mice.
Serum lipase, amylase, diamine oxidase (DAO) and endotoxins detection
Blood was drawn from the heart and centrifuged at 4000 rpm for 10 minutes to separate serum. Serum levels of lipase, amylase, DAO activity and endotoxins were measured using commercial kits according to the manufacturers’ protocols.
HE staining and TUNEL staining
Pancreas and intestinal segments were collected, fixed in 4% paraformaldehyde overnight at room temperature, and then routinely embedded in paraffin, sectioned at 5 μm thickness, and stained with HE to observe histopathological changes under a light microscope. Pathological scores were calculated in a double-blind manner as previously described.7,17,18 Pancreatic pathological scores were based on the degree of hemorrhage, edema, neutrophil infiltration, and necrosis. Intestinal damage severity was scored on a 0–5 scale (0, none; 1, mild; 2, moderate; 3, marked; 4, severe; 5, extremely severe).
For TUNEL staining, paraffin sections of intestinal segments were dewaxed, rehydrated, and incubated with proteinase K for 20 min at 37°C. After treatment with 3% H2O2 for 10 min at room temperature, sections were processed using a TUNEL assay kit following the manufacturer’s instructions. Cell nuclei were counterstained with DAPI (#ab285390, Abcam). TUNEL-positive cells were imaged using a fluorescence microscope (Nikon Eclipse NI-E; Nikon Corporation) and counted in three randomly selected fields per section to assess intestinal epithelial apoptosis.
Enzyme-linked immunosorbent assay (ELISA)
Serum concentrations of TNF-α, IL-1β, and IL-6 were determined using commercially available ELISA kits according to the manufacturers’ instructions.
Cell culture and transfection
MODE-K cells (the mouse intestinal epithelial cell line, #BFN608006456, Bluefbio. Inc., Shanghai, China) were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. After serum starvation for 12 h, exponentially growing cells (≈80% confluence) were transfected with adenoviral vectors overexpressing Mst1 (Ad-Mst1, MOI=100, customized by Hanbio Biotechnology, Shanghai, China) for 24 h. Cells transduced with control adenoviral vectors (Ad-Control, MOI=100) served as controls. All cells were then stimulated with 10 ng/mL TNF-α or equivalent saline in the presence or absence of bafilomycin A1 for an additional 24 h. At the end of the experiment, cells were collected for Western blot analysis.
Western blot analysis
Total protein was extracted from intestinal tissues or MODE-K cells, and protein concentrations were determined using the BCA method. Equal amounts of protein were separated by SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with primary antibodies, followed by HRP-conjugated secondary antibodies, and signals were detected using enhanced chemiluminescence. Immunoblots were analyzed using a gel imaging system (Bio-Rad, CA, USA). Expression levels of ZO-1, claudin-1, occludin, Mst1, p-Mst1, beclin-1, p62, LC3, cleaved caspase-3, Bcl-2, and Bax were measured. GAPDH was used as a loading control.
Statistical analysis
All data are expressed as mean ± standard deviation. Statistical differences were evaluated by one-way ANOVA followed by Dunnett’s test for multiple comparisons. Survival data were analyzed by the Kaplan Meier method with the log-rank test for multiple comparisons. A P value < 0.05 was considered statistically significant.
Result
Mst1 knockout improved the accumulative mortality and relieved intestinal barrier dysfunction of SAP mice
Mst1 knockout greatly reduced the mortality of SAP mice. As shown in Figure 1(a), the 5-day cumulative mortality in WT+SAP mice was approximately 75%, whereas that in Mst1-/-+SAP mice was about 55%, representing a marked reduction. WT+SAP mice exhibited severe pancreatic injury and intestinal barrier dysfunction, as evidenced by increased serum levels of lipase (Figure 1(b)), amylase (Figure 1(c)), DAO (Figure 1(d)), endotoxin (Figure 1(e)), and the inflammatory cytokines TNF-α, IL-1β, and IL-6 (Figure 1(i)–(k)). Histopathological examination of the pancreas revealed extensive widening of interlobular spaces, inflammatory cell infiltration, acinar cell necrosis, and hemorrhage (Figure 1(f)), leading to a significantly higher pancreatic pathological score in WT+SAP mice (Figure 1(g)). Similarly, ileocolic tissues from WT+SAP mice showed focal necrosis, epithelial cell detachment, basement membrane disruption, villous stroma broadening, edema, and congestion (Figure 1(f)), with a significantly increased intestinal pathological score (Figure 1(h)). Mst1 knockout improved cumulative survival and relieved intestinal barrier dysfunction of SAP mice. (A) SAP mice model was established via intraperitoneal injection of caerulein (50μg/kg per hour) for 10 times combining use of LPS (10 mg/kg) intraperitoneally injection at the last caerulein treatment. The mortality of mice was recorded every 24h for 5 days after the first injection of caerulein. Littermate mice were used in the survival study and each group contained 15 animals. Survival data was evaluated by the Kaplan Meier method followed by the log-rank test for multiple comparisons. **P < 0.01 vs. WT group, ##P < 0.01 vs. WT+SAP group. The contents of lipase (B), amylase (C), DAO (D), and endotoxin (E) in the serum were evaluated respectively. The pathohistological changes of pancreas and ileocolic mucosa were detected by HE staining (F). The bar represents 50μm. The pathological scores for pancreas (G) and ileocolic mucosa (H) were evaluated. The content of TNF-α (I), IL-1β (J), and IL-6 (K) in the serum were investigated. Data were means ±S.D., n= 10 mice/group. **P < 0.01 vs. WT group, ##P < 0.01 vs. WT+SAP group.
Mst1 knockout did not reduce serum lipase or amylase levels (Figure 1(b) and (c)) nor improve pancreatic histopathology or pathological scores in Mst1-/-+SAP mice (Figure 1(f) and (g)). However, Mst1 knockout significantly reduced serum DAO (Figure 1(d)) and endotoxin (Figure 1(e)) levels, ameliorated ileocolic histopathological changes (Figure 1(f) and (h)), and decreased serum levels of TNF-α, IL-1β, and IL-6 (Figure 1(i)–(k)). These results indicate that intestinal barrier dysfunction is a critical factor in the progression of SAP, and that Mst1 knockout significantly improves intestinal barrier function, thereby alleviating systemic symptoms and improving prognosis.
Mst1 knockout up-regulated tight junction proteins and alleviated apoptosis in the ileocolic mucosa tissue of SAP mice
We next examined the expression of the tight junction proteins ZO-1, claudin-1, and occludin in ileocolic mucosal tissue. As shown in Figure 2(a)–(d), the expression of all three proteins was markedly reduced in WT+SAP mice. In contrast, Mst1 knockout significantly upregulated these proteins in Mst1-/-+SAP mice. TUNEL staining revealed a significant increase in TUNEL-positive cells in the ileocolic mucosa of WT+SAP mice, whereas Mst1 knockout markedly reduced the number of apoptotic cells (Figure 2(e)). These findings suggest that reduced tight junction protein expression and increased apoptosis contribute to intestinal barrier dysfunction in WT+SAP mice, and that Mst1 knockout alleviates this dysfunction by restoring tight junction proteins and reducing apoptosis. Mst1 knockout up-regulated tight junction proteins and alleviated apoptosis in the ileocolic mucosa tissue of SAP mice. (A) The representative immunoblots of ZO-1, Claudin-1 and Occludin in the ileocolic mucosa tissue of SAP mice, GAPDH was used as a loading control. The levels of ZO-1 (B), Claudin-1 (C), Occludin (D) were analyzed (n=3 independent experiments). (E) Representative images of TUNEL staining (green) of ileocolic mucosa tissue section slices from mice of each group. The nuclei were counterstained with DAPI (blue), The bar represents 100 μm. Apoptosis index expressed as the percentage of TUNEL-positive cells (in green) over total nuclei determined by DAPI staining (n=20 fields per group). Data were means ±S.D., **P < 0.01 vs. WT group, ##P < 0.01 vs. WT+SAP group.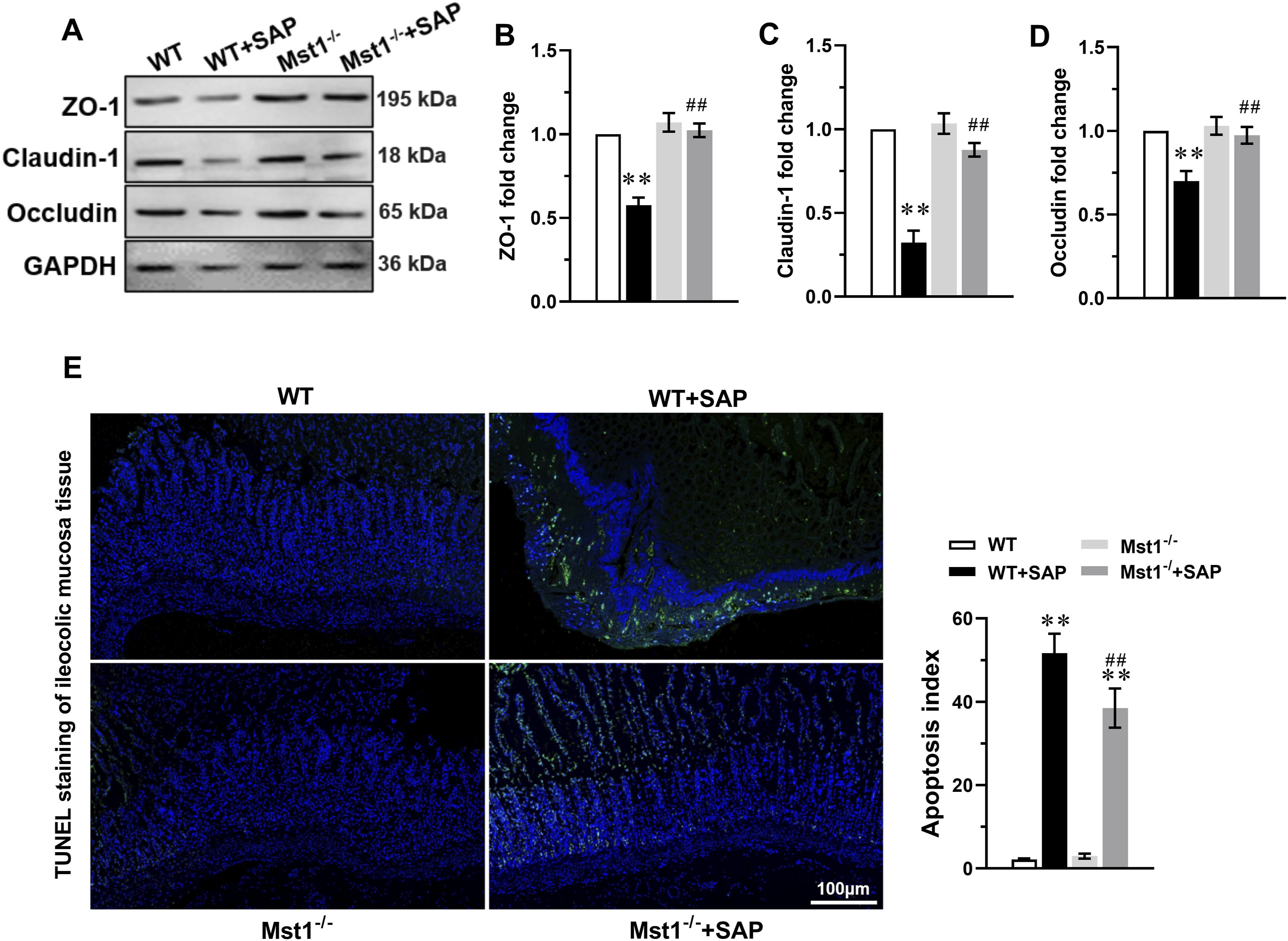
Mst1 knockout enhanced autophagy in the ileocolic mucosa of SAP mice
We further analyzed the expression of Mst1, p-Mst1, and autophagy-related proteins in ileocolic mucosal tissue. Mst1 and p-Mst1 levels were significantly elevated in WT+SAP mice (Figure 3(a)–(d)), accompanied by decreased beclin-1 and LC3-II expression and increased p62 expression (Figure 3(a), Figure 3(e)–(g) Mst1 knockout enhanced autophagy in the ileocolic mucosa tissue of SAP mice. (A) The representative immunoblots of Mst1, p-Mst1, Beclin-1, P62 and LC3 in the ileocolic mucosa tissue of SAP mice, GAPDH was used as a loading control. The levels of Mst1 (B), p-Mst1 (C), ratio of p-Mst1/Mst1 (D), Beclin-1 (E), P62 (F), and LC3 II (G) were analyzed. Data are expressed as mean ± S.D., n=3 independent experiments, **P < 0.01 vs. WT group, ##P < 0.01 vs. WT+SAP group.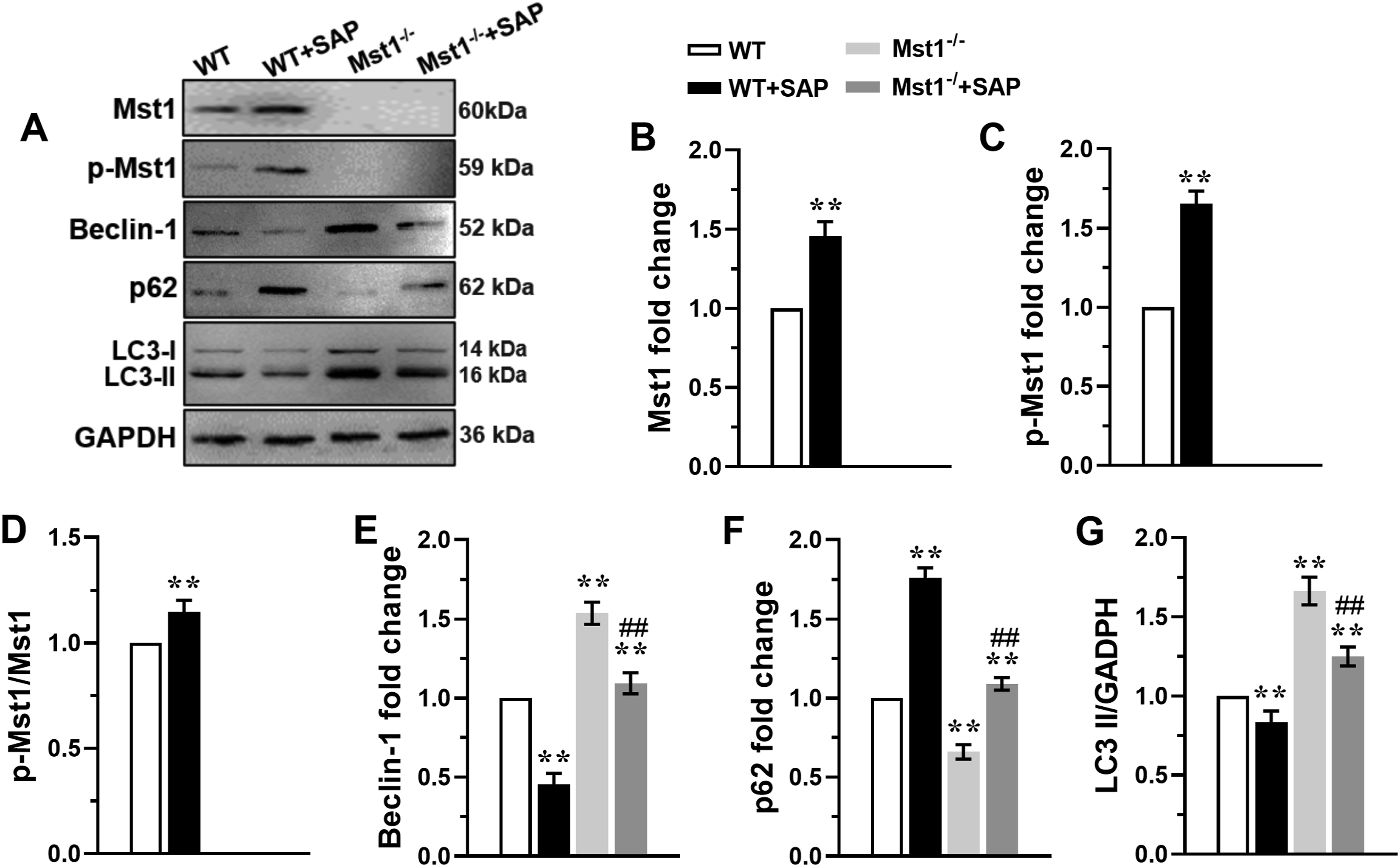
Mst1 overexpression inhibited autophagy and promoted apoptosis in MODE-K cells subjected to TNF-α stimulation
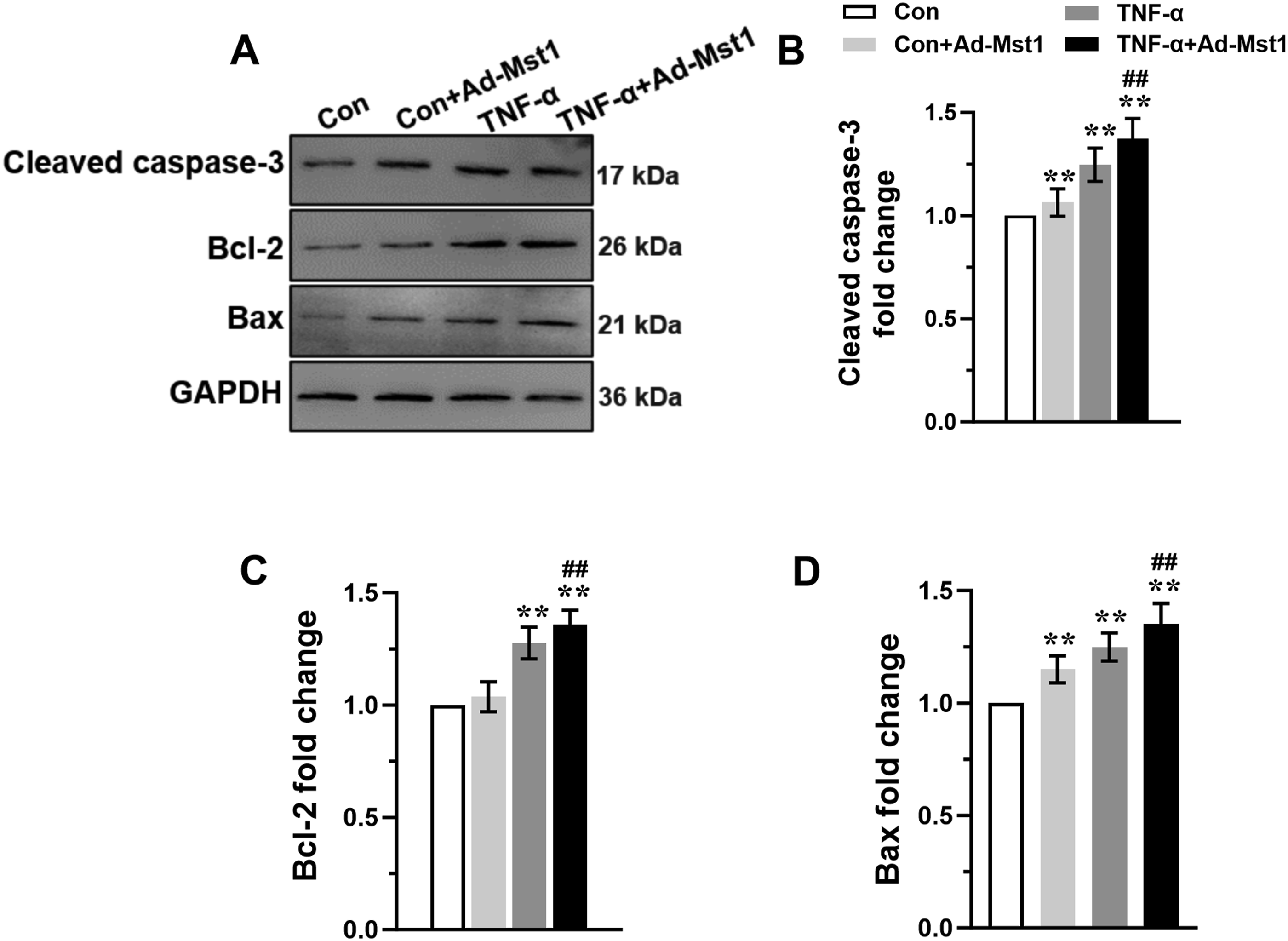
MODE-K cells transfected with Ad-Mst1 and stimulated with TNF-α (10 ng/mL) showed significantly increased Mst1, p-Mst1, and p62 expression, and decreased beclin-1 and LC3-II expression (Figure 4(a)–(f)). Furthermore, autophagic flux, assessed using the lysosomal inhibitor bafilomycin A1, was inhibited following Ad-Mst1 transfection, as shown by decreased LC3-II and increased p62 accumulation (Figure 4(g)–(i)). These data indicate that Mst1 overexpression blocks autophagic flux. Additionally, Ad-Mst1 transfection increased the expression of cleaved caspase-3, Bcl-2, and Bax compared with the TNF-α alone group (Figure 5(a)–(d)). Collectively, these results demonstrate that Mst1 overexpression inhibits autophagic flux and promotes apoptosis in TNF-α-stimulated MODE-K cells. Mst1 overexpression inhibited autophagy in MODE-K cells subjected to TNF-α stimulation. Exponentially growing MODE-K cells transfected with Ad-Mst1 or Ad-Control were stimulated with 10 ng/ml of TNF-α to detect the expression of autophagy related molecules. (A) The representative immunoblots of Mst1, p-Mst1, Beclin-1, P62 and LC3 in MODE-K cells. GAPDH was used as a loading control. The levels of Mst1 (B), p-Mst1 (C), Beclin-1 (D), P62 (E), and LC3 II (F) were analyzed. (G) Representative blots and analysis of p62 and LC3 in the absence or presence of bafilomycin A1 in MODE-K cells stimulated with 10 ng/ml of TNF-α. Data are expressed as mean ± S.D., n=3 independent experiments, **P < 0.01 vs. Con group, ##P < 0.01 vs. Con+ Ad-Mst1 group, && P < 0.01 vs. TNF-α group. Mst1 overexpression promoted apoptosis in MODE-K cells subjected to TNF-α stimulation. The expression of apoptosis related molecules in MODE-K cells transfected with Ad-Mst1 or Ad-Control following stimulation with 10 ng/ml of TNF-α were investigated. (A) The representative immunoblots of Cleaved caspase-3, bcl-2, and Bax in MODE-K cells. GAPDH was used as a loading control. The levels of Cleaved caspase-3 (B), bcl-2 (C), and Bax (D) were analyzed. Data are expressed as mean ± S.D., n=3 independent experiments, **P < 0.01 vs. Con group, ##P < 0.01 vs. TNF-α group.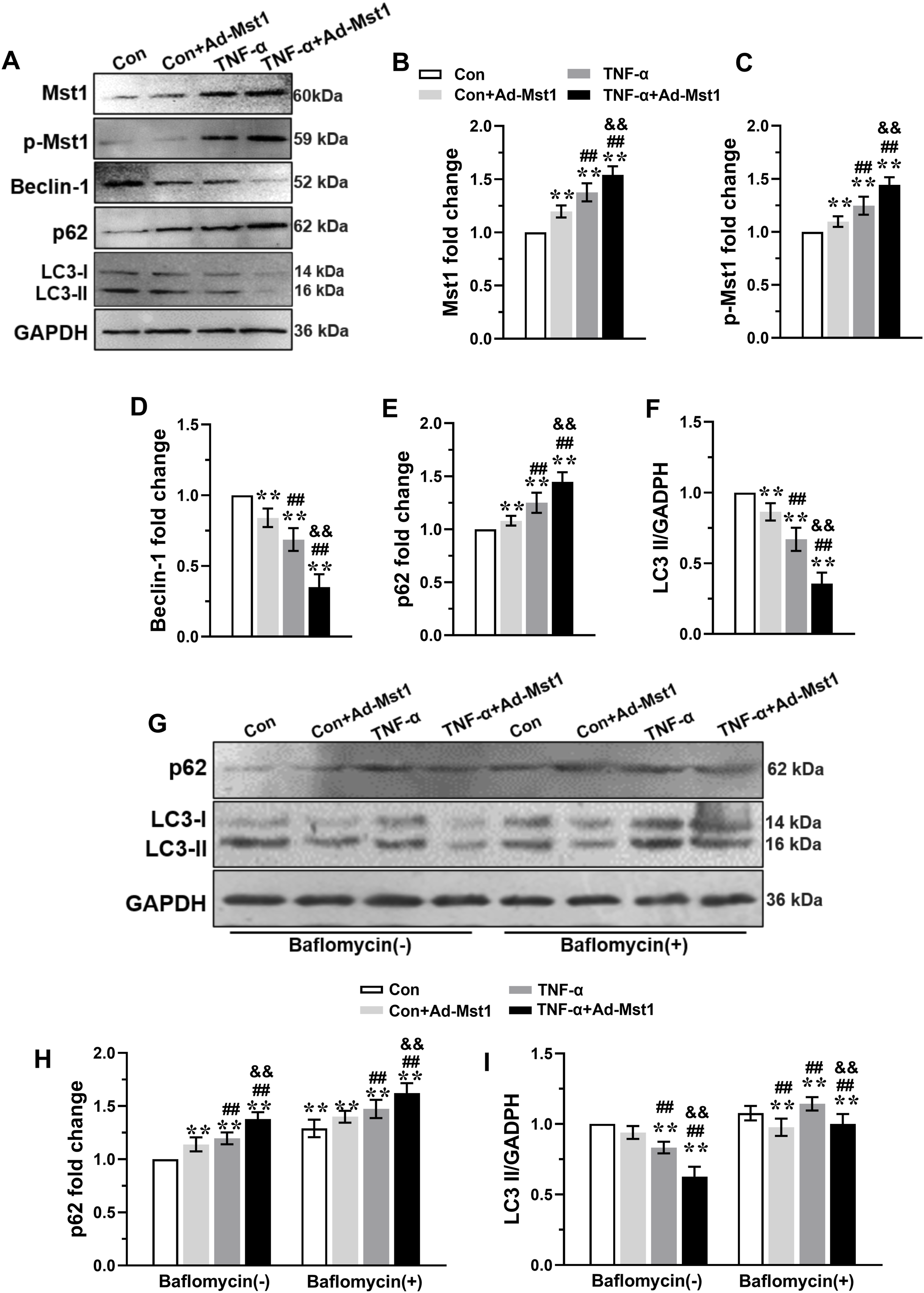
Discussion
Our study presents the novel finding that Mst1 plays an important role in SAP-related intestinal barrier dysfunction. We found that Mst1 and phosphorylated Mst1 levels are markedly increased, leading to autophagy inhibition and apoptosis promotion in intestinal mucosal cells, thereby aggravating intestinal barrier dysfunction in SAP mice. Conversely, Mst1 knockout enhanced autophagy, suppressed apoptosis, upregulated tight junction proteins, improved intestinal barrier function, and reduced SAP-associated mortality.
The intestinal barrier is a complex system comprising mechanical, immune, chemical, and biological components that prevent bacterial translocation and maintain intestinal homeostasis. Numerous studies have shown that excessive apoptosis and suppressed autophagy of intestinal epithelial cells contribute to intestinal barrier impairment during SAP, altering permeability and integrity, and subsequently triggering SIRS and MODS. In this study, the SAP model was successfully established by intraperitoneal injection of caerulein (50 μg/kg per hour for 10 times) combined with LPS (10 mg/kg) at the last caerulein injection. WT mice developed severe acute pancreatitis, as evidenced by pronounced pancreatic histopathological changes and elevated serum lipase and amylase levels. Intestinal barrier dysfunction was also observed in WT+SAP mice, with severe histopathological alterations including focal necrosis, epithelial cell detachment, basement membrane disruption, villous stroma broadening, edema, and congestion. Moreover, the expression of ZO-1, claudin-1, and occludin was markedly reduced in the ileocolic mucosa of WT+SAP mice. These changes led to increased intestinal permeability and elevated serum levels of DAO, endotoxin, and the inflammatory cytokines TNF-α, IL-1β, and IL-6. Importantly, increased apoptosis and impaired autophagy were detected in intestinal epithelial cells of WT+SAP mice, representing a key mechanism underlying the observed intestinal barrier dysfunction.
The intestinal microenvironment during SAP is highly complex, involving dynamic interactions among intestinal epithelial cells, immune cells, the gut microbiota, and a network of inflammatory mediators.19–21 In SAP, excessive systemic inflammation and ischemia-reperfusion injury compromise intestinal epithelial integrity, leading to increased permeability and bacterial translocation. In turn, infiltrating immune cells such as neutrophils and macrophages release pro-inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6), which further damage intestinal epithelial cells and disrupt tight junction proteins. Simultaneously, the gut microbiota undergoes dysbiosis, characterized by a reduction in beneficial commensals and overgrowth of pathogenic bacteria, which exacerbates local and systemic inflammation via pattern recognition receptor activation. These interconnected factors create a vicious cycle that amplifies intestinal barrier dysfunction and promotes the progression of SAP-related SIRS and MODS. Within this complex microenvironment, Mst1 appears to act as a key intracellular mediator, responding to inflammatory and stress signals to tip the balance between autophagy and apoptosis in intestinal epithelial cells. Therefore, understanding these multi-dimensional interactions is essential for developing targeted therapies that preserve intestinal barrier function in SAP patients.
Mst1 exerts bidirectional regulatory effects on autophagy and apoptosis. This dual functionality is essential for maintaining cardiovascular health by balancing cell survival and the removal of harmful cellular components, thereby preventing the progression of atherosclerosis, myocardial infarction, and heart failure.9,10,12,13,22 However, its role in SAP-induced intestinal barrier dysfunction has remained largely unexplored. In this study, we observed a significant upregulation of Mst1 and its phosphorylated form, pMst1, in the intestinal tissues of WT+SAP mice. This increase in Mst1 expression may be a critical factor leading to increased apoptosis and decreased autophagy in intestinal epithelial cells. The precise molecular mechanism by which Mst1 bidirectionally regulates autophagy and apoptosis in intestinal epithelial cells during SAP may involve the phosphorylation of beclin-1 and the regulation of the Bcl-2/Bax-caspase-3 axis, as previously reported in cardiac studies. 9 However, whether similar pathways operate in intestinal epithelial cells requires further validation. Furthermore, we also found that Mst1 overexpression inhibited autophagic flux and promoted apoptosis in MODE-K cells subjected to TNF-α stimulation, further supporting the role of Mst1 in modulating the balance between these two cellular processes in the context of SAP-induced intestinal barrier dysfunction.
From a clinical perspective, Mst1 inhibition could be achieved using small-molecule inhibitors (e.g., XMU-MP-1) or gene therapy approaches. These strategies may help preserve intestinal barrier function, reduce bacterial translocation, and improve outcomes in SAP patients. Future clinical trials are warranted to test the safety and efficacy of Mst1-targeted therapies in SAP-associated intestinal dysfunction. Clinically, intestinal barrier dysfunction is a key determinant of disease progression and prognosis in SAP patients. Our findings suggest that Mst1 may serve as a potential biomarker for early identification of SAP patients at high risk of intestinal barrier injury. Furthermore, pharmacological inhibition of Mst1 could represent a novel therapeutic strategy to restore intestinal integrity and reduce systemic inflammation in SAP.
Although our study demonstrates that Mst1 plays an important role in SAP-related intestinal barrier dysfunction by bidirectionally regulating autophagy and apoptosis, several limitations should be addressed in future research. First, the exact mechanism by which Mst1 mediates autophagy and apoptosis in intestinal epithelial cells during SAP requires further clarification. Second, the upstream signals responsible for Mst1 and p-Mst1 upregulation during SAP remain unidentified. Potential candidates include inflammatory cytokines (e.g., TNF-α, IL-6) and oxidative stress, which are known to activate Hippo signaling. Future studies should investigate whether TLR4/NF-κB or reactive oxygen species pathways are involved in Mst1 activation during SAP. Third, this study was conducted solely in mouse models and a murine intestinal epithelial cell line. Therefore, the translational relevance of our findings to human SAP patients requires further validation in large animal models and clinical cohort studies. All these limitations represent interesting avenues for future research.
Conclusion
Mst1 plays a critical role in SAP-induced intestinal barrier dysfunction by inhibiting autophagy and promoting apoptosis in intestinal epithelial cells. Mst1 knockout enhances autophagy, reduces apoptosis, upregulates tight junction proteins, and improves survival in SAP mice. Mst1 may serve as a novel therapeutic target for preventing intestinal barrier failure in SAP patients.
Footnotes
Acknowledgements
Thank Home for Researchers editorial team (www. Home-for-researchers.com) for providing language editorial service. All authors thank the editor and the reviewers for providing constructive input and assistance to improve this paper.
Ethical considerations
All animal experiments were strictly performed in accordance with the guidelines for the Care and Use of Laboratory Animals issued by the Laboratory Animal Welfare and Ethics Committee of Northwest University (Xi’an), the National Research Council’s Guide for the Care and Use of Laboratory Animals, and the ARRIVE guidelines. This study was formally approved by the Laboratory Animal Welfare and Ethics Committee of Northwest University (Xi’an) (Approval No.: NWU-LAEC-XA-2023-071). All procedures were designed to minimize animal suffering and discomfort, and every effort was made to reduce the number of animals used, in strict adherence to the 3R principles.
Author contributions
Rong Zhang: Conceptualization, Writing – original draft. Ziwen Liu: Data curation, Investigation. Tao Lin: Methodology, Validation. Rui Shen: Formal analysis, Software. Chuqi Xiang: Resources, Visualization. Pengtao Zhao: Supervision, Funding acquisition, Writing – review & editing. Wang Lu: Project administration, Funding acquisition, Writing – review & editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the following grants: National Natural Science Foundation of China (No. 82170094); Science and Technology Research and Development Program of Shaanxi Province, China (No. 2020SF-220, 2022SF-462).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The original data of the study are included in this article, and further inquiries can be directed to the corresponding author on reasonable request.