
Abstract
Pseudohypoparathyroidism (PHP) is a heterogeneous group of disorders defined by resistance to parathyroid hormone (PTH), typically stemming from GNAS locus abnormalities. This report describes an unusual case of a 32-year-old Han Chinese male with sporadic PHP Type 1B (PHP1B) who presented with paroxysmal numbness and significant hypokalemia. Despite elevated PTH levels, the patient maintained normocalcemia and showed no evidence of globus pallidus calcification on brain imaging. Diagnosis was confirmed using methylation-specific multiplex ligation-dependent probe amplification, which identified characteristic GNAS imprinting defects: a gain of methylation at the NESP55 exon and loss of methylation at the AS, XL, and A/B exons. With a normal copy number, no detectable GNAS mutations, and a negative family history, the case was classified as sporadic PHP1B. This rare presentation highlights the broad phenotypic spectrum of PHP and emphasizes the clinical importance of monitoring diverse electrolyte disturbances, such as hypokalemia, even when calcium levels appear normal.
Introduction
Pseudohypoparathyroidism (PHP) is a rare, sporadic or inherited genetic disorder that is classified into several distinct subtypes, including types Ia, Ib, Ic, and type II, based on differences in pathogenesis and clinical phenotype. Current epidemiological studies report an estimated incidence ranging from approximately 0.34 per 100,000 to 1.1 per 100,000 individuals.1,2 PHP represents a clinical syndrome characterized by resistance of peripheral target tissues to parathyroid hormone (PTH). However, PHP shares biochemical features with hypoparathyroidism, circulating PTH levels are markedly elevated. Despite increased PTH secretion, impaired hormone responsiveness in target organs leads to hypocalcemia and hyperphosphatemia, often accompanied by reduced serum levels of 1,25(OH)2D. The predominant clinical manifestations are associated with disturbances in calcium and phosphate homeostasis. In addition, patients with PHP type Ia or Ic, as well as a subset of patients with PHP1B, may exhibit features of Albright hereditary osteodystrophy (AHO), including short stature, obesity, a round face, a short neck, brachydactyly, ectopic calcifications, and variable degrees of intellectual impairment. 3
PHP is typically classified into type 1 and type 2 based on the urinary cyclic adenosine monophosphate (cAMP) response to exogenous PTH stimulation. 4 Type 1 PHP is further subdivided based on the presence or absence of Albright hereditary osteodystrophy (AHO) and the underlying molecular defects. According to the EuroPHP international consensus, 5 PHP-Ia is caused by heterozygous inactivating mutations in the GNAS gene on the maternal allele, leading to reduced Gsα protein expression or function; these patients typically exhibit AHO features and multihormone resistance (PTH, TSH, GnRH). PHP-Ic is characterized by GNAS mutations that impair receptor coupling while preserving basal Gsα activity, presenting with a clinical phenotype similar to PHP-Ia, including AHO and multihormone resistance. In contrast, PHP-Ib arises from methylation defects at the differentially methylated regions (DMRs) of the GNAS locus, resulting in isolated renal resistance to PTH; AHO features are generally absent, although partial TSH resistance has been reported.6–8 PHP type 2 is distinguished by a normal cAMP response but absent phosphaturic response to exogenous PTH. 4 PHP is commonly associated with disturbances in electrolyte homeostasis. Several studies have reported that PHP, particularly PHP1B, may present with concomitant hypocalcemia and hypokalemia.9–11 However, sporadic cases of PHP1B accompanied by persistent hypokalemia in the setting of normal serum calcium have rarely been documented. The reporting of this case conforms to the CARE guidelines. 12 The present report describes a male patient with normocalcemic PHP complicated by marked hypokalemia and examines the potential underlying pathophysiological mechanisms.
Case presentation
On May 8, 2024, a 32-year-old Han Chinese male patient was admitted to the Department of Endocrinology at Qilu Hospital, Shandong University, due to recurrent numbness involving the face and limbs for a duration of 2 years. The initial symptoms appeared 2 years earlier and consisted of episodic numbness affecting the face, upper limbs, and chest. These episodes were frequently triggered by alcohol consumption and resolved spontaneously, leading the patient to disregard them initially.
By June 2023, the frequency and severity of the episodes increased, with emotional stress, sleep deprivation, and physical fatigue identified as frequent precipitating factors for the patient. More severe attacks were accompanied by perioral numbness, muscle spasms, and carpopedal spasm. From June 2023, the episodes occurred with a frequency of approximately 2–3 times per week, each lasting 5–30 min, and resolved spontaneously without intervention.
On June 3, 2023, the patient was admitted to the Department of Endocrinology at Liaocheng People’s Hospital, Shandong Province, due to worsening episodic numbness, perioral numbness, muscle spasms, and carpopedal spasm. The patient reported no prior use of diuretic agents, calcium supplements, or vitamin D preparations. Laboratory evaluation revealed hypokalemia (serum K+ 2.87 mmol/L, ref. 3.5–5.0), elevated CO2 (35.4 mmol/L, ref. 18.0–28.0), and calcium at 2.28 mmol/L(ref. 2.11–2.52). The elevated CO₂ (35.4 mmol/L) in the setting of hypokalemia and renal potassium wasting indicated a mild metabolic alkalosis, which normalized to 26.1 mmol/L on the current admission after potassium repletion and spironolactone therapy—a pattern typical of hypokalemia-associated alkalosis rather than a primary tubular defect.
Assessment of cortisol circadian rhythm and 24-h urinary cortisol excretion yielded normal results, as evaluated the renin–angiotensin–aldosterone system (RAAS). Although overnight dexamethasone suppression test and midnight salivary cortisol were not performed, the normal 24-h urinary free cortisol and preserved cortisol diurnal variation, together with the absence of clinical features of hypercortisolism, effectively excluded Cushing syndrome as a cause of hypokalemia. Thyroid function tests showed low FT4 and elevated TSH.
During hypokalemia (serum K+ 3.03 mmol/L), 24-h urinary potassium excretion was 92.4 mmol/24 h. According to established physiological criteria (urinary K+ >20 mmol/24 h in the setting of serum K+ <3.5 mmol/L), this level is inappropriately high and indicates renal potassium wasting. This was further supported by a urine K+/creatinine ratio of 8.1 mmol/mmol (reference <2.5 in hypokalemia). A diagnosis of hypokalemia secondary to renal potassium loss was established, and potassium supplementation was administered, resulting in partial symptomatic improvement.
For further evaluation and management, the patient was admitted to the current department on May 8, 2024. At admission, the patient measured 184 cm in height and weighed 100 kg, with a calculated body mass index (BMI) of 29.5 kg/m², placing him in the overweight to borderline obesity range. Apart from this elevated BMI, no other AHO features (such as brachydactyly, round face, short neck, or subcutaneous ossifications) were present, and his overall physical development was otherwise unremarkable. No goiter was observed, and cardiopulmonary and abdominal examinations revealed no abnormalities. Muscle strength and tone were preserved in all extremities, without evidence of muscle atrophy or joint deformity. No family history of PHP was reported, and both parents were phenotypically normal with average stature.
Laboratory investigations revealed elevated serum PTH levels in the presence of normal serum calcium, serum phosphorus, and urinary calcium excretion (Table 1). The serum 25-hydroxyvitamin D3 level was decreased (17.54 ng/mL, reference range: 30–100 ng/mL), indicating vitamin D deficiency. Although vitamin D deficiency can affect calcium metabolism, the patient was not given cholecalciferol or ergocalciferol supplementation; instead, active vitamin D (calcitriol 0.25 μg once daily) was initiated to target the underlying PTH resistance. Persistent hypokalemia with renal potassium loss was accompanied by elevated direct renin concentration and normal aldosterone levels. Serum magnesium, creatinine, and carbon dioxide combining power remained within normal ranges. Thyroid function evaluation was consistent with subclinical hypothyroidism, characterized by elevated TSH levels and normal thyroxine levels, along with increased thyroid peroxidase antibody levels.
Baseline laboratory findings of the patient.
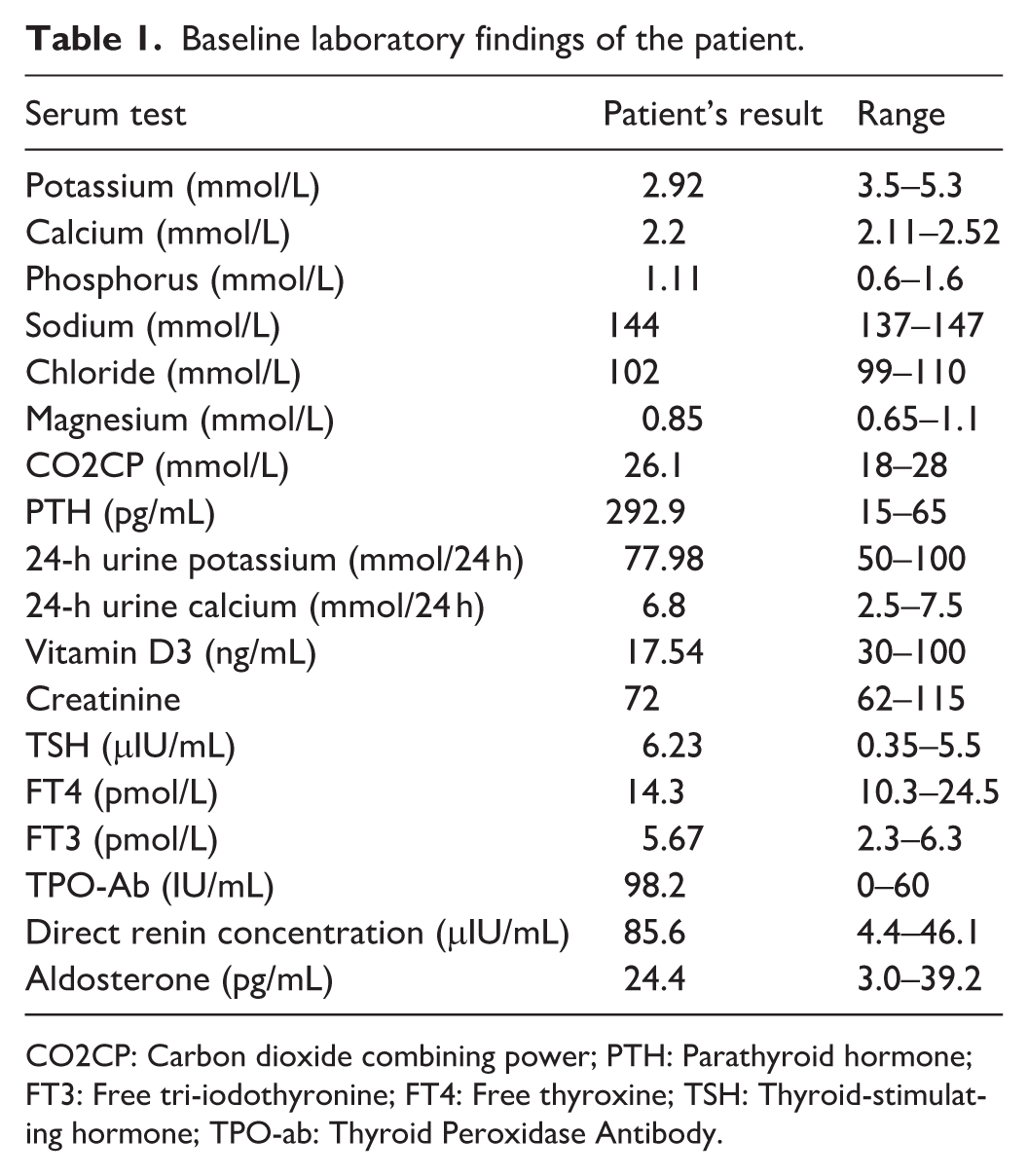
CO2CP: Carbon dioxide combining power; PTH: Parathyroid hormone; FT3: Free tri-iodothyronine; FT4: Free thyroxine; TSH: Thyroid-stimulating hormone; TPO-ab: Thyroid Peroxidase Antibody.
Brain computed tomography revealed no evidence of globus pallidus calcification. Radiographic examinations of the skull, pelvis, hands, and lower limbs demonstrated no significant skeletal abnormalities. Cervical ultrasonography identified a hypoechoic nodule located posterior to the mid-to-lower portion of the right thyroid lobe, suggestive of parathyroid origin. Parathyroid scintigraphy demonstrated nodular lesions with mild radiotracer uptake posterior to both lower poles and the mid-right thyroid lobe, raising suspicion for parathyroid adenoma or hyperplasia. Bone mineral density was within the normal range.
Whole-exome sequencing was performed after informed consent was obtained from the patient and family members. Sequence analysis did not identify pathogenic variants in the GNAS gene. Given that copy number variation analysis by next-generation sequencing may miss small deletions or duplications, multiplex ligation-dependent probe amplification (MLAP) was subsequently performed. The MLPA analysis demonstrated gain of methylation at the NESP55 exon and loss of methylation at the AS, XL, and A/B exons (Figure 1).
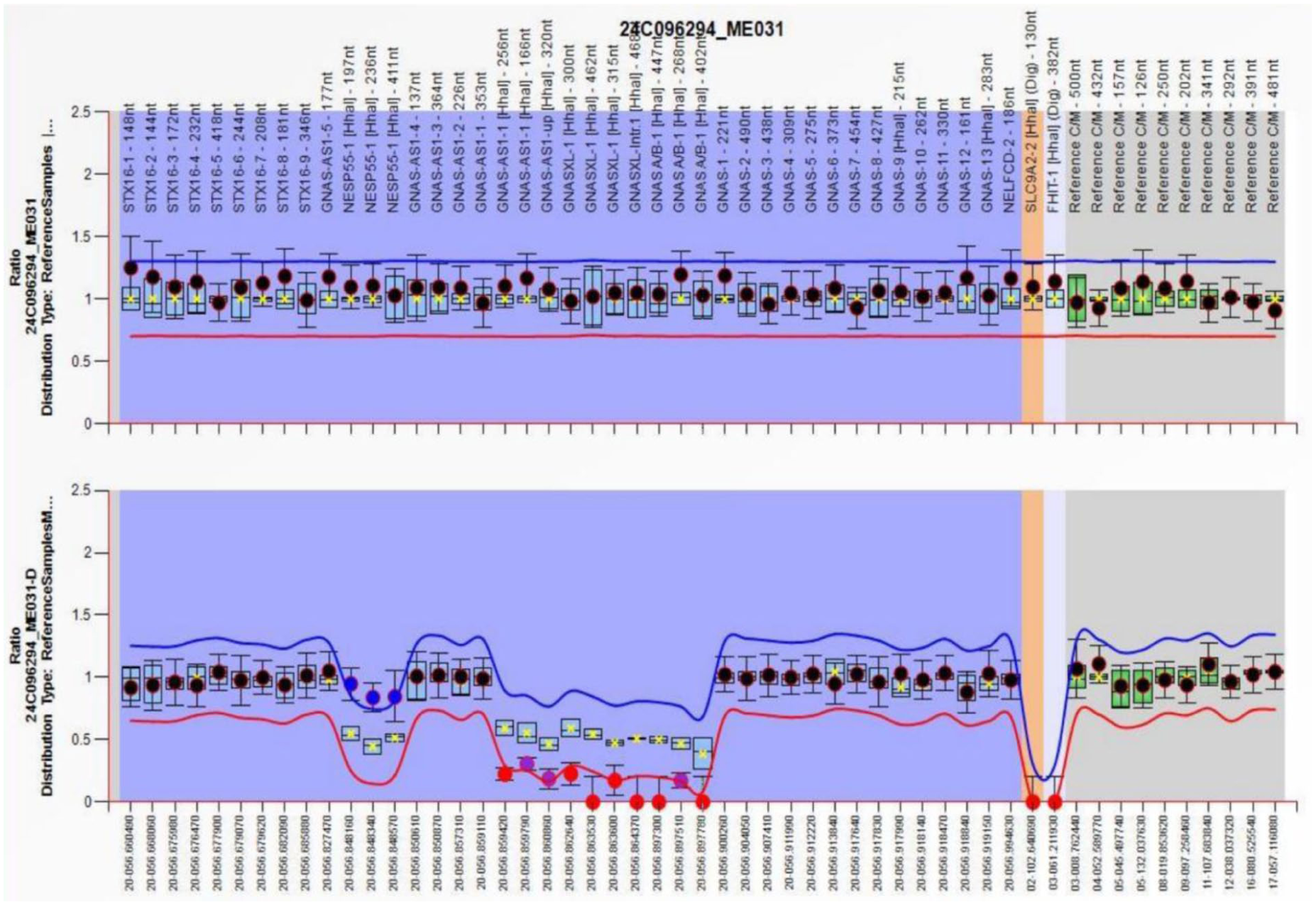
MS-MLPA analysis of the GNAS locus.
Several diagnostic challenges were encountered. First, the patient’s normocalcemia initially diverted attention away from PHP, as hypocalcemia is traditionally considered a hallmark. Second, the prominent hypokalemia with renal potassium wasting raised suspicion of tubulopathies such as Gitelman or Bartter syndrome, leading to targeted genetic testing that excluded these conditions. Third, the absence of AHO features and basal ganglia calcification further obscured the diagnosis of PHP1B. Only after whole-exome sequencing and GNAS methylation-specific MLPA was the correct diagnosis established, highlighting the importance of considering PHP1B even in normocalcemic patients with unexplained hypokalemia.
The diagnosis of sporadic Pseudohypoparathyroidism Type 1B (PHP1B) was confirmed based on the absence of a family history of PHP. 5 Medical management consisted of oral potassium chloride 2 g three times daily, spironolactone 20 mg once daily, and calcitriol 0.25 μg once daily. Although serum aldosterone was within the normal range, spironolactone was added empirically to enhance renal potassium conservation, given the persistent renal potassium wasting and elevated renin concentration. This approach was based on the hypothesis that potassium supplementation might induce secondary hyperaldosteronism and that spironolactone could provide additional kaliuretic blockade at the distal tubule.
A follow-up evaluation 3 months later demonstrated marked clinical improvement. Laboratory assessment showed normalization of serum potassium to 3.80 mmol/L and maintenance of serum calcium at 2.22 mmol/L. Serum PTH levels decreased significantly to 189 pg/mL following treatment. No adverse effects or unanticipated events were reported during the follow-up period. From the patient’s perspective, the treatment was well tolerated and resulted in complete relief of previous symptoms, with no impact on his quality of life or occupational activities (Figure 2).

Clinical timeline of the patient from initial presentation to follow-up.
Discussion and conclusion
The present report describes a patient with normocalcemic PHP accompanied by chronic hypokalemia. Clinical evaluation revealed no other phenotypic features of AHO besides borderline obesity. Thyroid function assessment was consistent with subclinical hypothyroidism. It is important to note that the patient’s subclinical hypothyroidism was accompanied by elevated thyroid peroxidase antibodies (TPO-Ab 98.2 IU/mL, reference 0–60), which strongly suggests coexistent Hashimoto’s autoimmune thyroiditis. While PHP1B can be associated with mild TSH resistance, the presence of positive thyroid autoantibodies makes autoimmune disease the more likely primary cause of the TSH elevation in this case. Therefore, we interpret the patient’s thyroid status as most consistent with two concurrent diagnoses: autoimmune subclinical hypothyroidism (Hashimoto’s thyroiditis) and PHP1B. and CT scanning showed no evidence of globus pallidus calcification. Based on biochemical findings and molecular analyses, a diagnosis of PHP1B was established. PHP is classically characterized by hypocalcemia. Although several recent reports have described cases of PHP1B presenting with both hypocalcemia and hypokalemia, no cases have been documented in which normocalcemic PHP coexists with hypokalemia.
To the best of our knowledge, this is the first reported case of PHP with normal serum calcium combined with hypokalemia. The physiological actions of PTH are mediated through binding to its receptor, which is coupled to Gsα proteins. Receptor activation stimulates adenylyl cyclase, increasing intracellular cAMP levels. As a second messenger, cAMP activates a cascade of protein kinases that ultimately mediate the diverse cellular effects of PTH. 13 The pathophysiology of PHP1B arises from genetic or epigenetic alterations in the GNAS gene, leading to reduced expression or impaired function of Gs proteins. Several mechanisms underlying epigenetic dysregulation have been identified, including stable alterations in DNA methylation patterns, posttranslational histone modifications, and regulatory effects mediated by non-coding RNAs. 14 These abnormalities lead to resistance to PTH, which typically manifests clinically as hypocalcemia and hyperphosphatemia, thereby contributing to symptoms such as muscle cramps and tetany.
The patient described was diagnosed with PHP1B based on MLPA findings. However, laboratory evaluation did not reveal hypocalcemia or hyperphosphatemia, and cranial CT showed no evidence of globus pallidus calcification. Similar observations have been reported in previous studies, indicating that some patients with PHP, particularly PHP1A, may present with normocalcemia.15–18 One proposed explanation for preserved serum calcium levels involves the maintained responsiveness of bone tissue to PTH. Ish-Shalom demonstrated that osteoblastic cells derived from patients with PHP1A retain an intact PTH receptor-coupled adenylyl cyclase signaling pathway in vitro, despite marked impairment of hormone-responsive adenylyl cyclase activity in other target tissues, particularly the kidney. 19 The methylation pattern identified in our patient—loss of methylation at the A/B, XL, and AS exons together with gain of methylation at NESP55—is the hallmark epigenetic signature of sporadic PHP1B. 9 This imprinting defect leads to reduced Gsα expression primarily in the renal proximal tubule, accounting for isolated renal PTH resistance. However, the patient’s normocalcemia may be explained by preserved PTH responsiveness in bone, as the methylation abnormalities do not equally affect all Gsα-expressing tissues. The absence of AHO features is consistent with PHP1B, where methylation defects spare the maternal Gsα promoter in most extrarenal tissues. Thus, the specific DMR pattern not only confirms the diagnosis but also helps explain the atypical normocalcemic presentation and the lack of brain calcifications. An additional explanation for the normocalcemic presentation in this patient may be a relatively mild degree of PTH resistance. Hyperparathyroidism in the presence of normocalcemia is generally considered an early and mild manifestation of PTH resistance. In a cohort of 19 children with PHP1B caused by maternally inherited STX16 deletions, the severity of PTH resistance increased with age, whereas TSH resistance showed no age-related association. 20 These findings are supported by another study demonstrating that older patients with PHP exhibit higher PTH and serum phosphorus levels, even in the absence of hypocalcemia. 21 Therefore, regular monitoring with advancing age is recommended to allow timely identification of overt hypocalcemia before the development of clinically significant manifestations. It is also important to recognize that normocalcemia in PHP1B does not represent an atypical variant separate from classic disease, but rather may reflect an early or partial stage of PTH resistance. As suggested by previous studies, the severity of PTH resistance in PHP1B can increase with age, and normocalcemic hyperparathyroidism may precede the development of overt hypocalcemia. Therefore, presenting this case as normocalcemic PHP1B should not be interpreted as a distinctly novel entity, but as part of a continuous clinical spectrum. This perspective has direct implications for clinical practice: in patients with suggestive features (e.g. unexplained hypokalemia, elevated PTH with normal calcium), clinicians should maintain a high index of suspicion for PHP even in the absence of hypocalcemia. Moreover, regular long-term follow-up is essential to detect potential progression to hypocalcemia, allowing timely intervention before serious complications arise.
Several studies have found that PHP can present with significant hypokalemia. Zhang et al. 22 reported a case involving a 27-year-old male patient with PHP1B coexisting with Gitelman syndrome. Yang et al. and Huang et al. also reported cases of PHP complicated by hypokalemia.9,23 To establish a structured diagnostic algorithm for hypokalemia in suspected PHP1B, we propose the following stepwise approach: (1) biochemical profiling confirming PTH resistance (elevated PTH with normal or low serum calcium) and renal potassium wasting (24 h urinary K+ >25–30 mmol/L despite hypokalemia); (2) exclusion of common renal tubular disorders (e.g. Gitelman/Bartter syndromes) via targeted genetic testing (SLC12A3, SLC12A1, KCNJ1, BSND, CLCNKB, CLCNKA, CASR); (3) if negative, proceed to whole-exome sequencing to identify other potential causes; (4) confirm PHP1B by GNAS methylation-specific MLPA, as recommended by Zhang and colleagues. 24 This systematic exclusion, combined with characteristic neuroimaging (lack of basal ganglia calcification) and absence of AHO features, supports the diagnosis of sporadic PHP1B as the underlying etiology of hypokalemia in this case.
The mechanism underlying hypokalemia in PHP1B is likely multifactorial. The traditional explanation focuses on impaired Gsα/cAMP signaling due to GNAS epigenetic defects, which reduces adenylyl cyclase activity in the renal proximal tubule. Attenuation of cAMP signaling may suppress potassium channel activity in the apical membrane of the thick ascending limb of the loop of Henle, disrupting potassium recycling and reducing intraluminal K+ concentration, which in turn inhibits the Na+-K+-2Cl– cotransporter (NKCC2), leading to sodium and potassium wasting. 24 However, emerging evidence suggests additional contributory factors. First, secondary hyperaldosteronism—possibly driven by renal salt wasting or mild volume contraction—can enhance distal nephron potassium secretion via epithelial sodium channels (ENaC).9,23 Second, a direct tubular defect in potassium handling, independent of aldosterone, has been proposed in some PHP patients, 22 though no pathogenic variants in Bartter/Gitelman syndrome genes were identified in our case. Third, resistance to PTH may also affect the distal convoluted tubule, where PTH normally promotes potassium reabsorption; loss of this effect could exacerbate renal potassium loss. Recent work emphasizes that hypokalemia in PHP1B arises from a combination of impaired Gsα/cAMP signaling, altered renal potassium channel activity, secondary hyperaldosteronism, and possible generalized tubular dysfunction. 24 While the cAMP-mediated mechanism remains a core pathway, the integrated model above better reflects the complex pathophysiology observed in clinical practice. Moreover, In our patient, direct renin concentration was elevated while aldosterone levels remained normal, a discordance that challenges the classical aldosterone-centric model of distal K+ secretion. One possible explanation is that increased distal sodium delivery (resulting from NKCC2 dysfunction) stimulates flow-dependent K+ secretion via high-conductance BK channels in the connecting tubule and collecting duct, a pathway that operates independently of aldosterone.25,26 Alternatively, a direct tubular defect in K+ handling or subtle, unmeasured aldosterone sensitivity cannot be entirely excluded. We acknowledge this renin–aldosterone dissociation as an unresolved but potentially informative finding that warrants further investigation.
In summary, this case report describes a rare presentation of sporadic normocalcemic PHP1B complicated by persistent hypokalemia. The diagnosis was confirmed by MLPA, which revealed a characteristic GNAS imprinting defect without pathogenic mutations in hypokalemia-related genes. This characteristic epigenetic profile, in the absence of coding mutations, is considered diagnostic of sporadic PHP1B. Further studies are required to elucidate the mechanisms underlying hypokalemia in PHP. The findings also highlight the phenotypic heterogeneity of PHP1B and emphasize the importance of evaluating electrolyte disturbances even when classic hypocalcemia is absent. The short-term prognosis in this patient was favorable with normalization of potassium and improvement of PTH levels. Long-term monitoring for emerging hypocalcemia and electrolyte disturbances is recommended, as PTH resistance may progress with age.
Footnotes
List of abbreviations
Abbreviation Full term
AHO Albright hereditary osteodystrophy
BMI Body mass index
cAMP Cyclic adenosine monophosphate
CO₂CP Carbon dioxide combining power
CT Computed tomography
DMR Differentially methylated region
ENaC Epithelial sodium channel
FT₃ Free tri-iodothyronine
FT₄ Free thyroxine
GNAS GNAS complex locus (encoding Gsα protein)
MLPA Multiplex ligation-dependent probe amplification
MS-MLPA Methylation-specific multiplex ligation-dependent probe amplification
NKCC2 Sodium-potassium-chloride cotransporter type 2
PHP Pseudohypoparathyroidism
PHP1B Pseudohypoparathyroidism type 1B
PTH Parathyroid hormone
RAAS Renin-angiotensin-aldosterone system
TSH Thyroid-stimulating hormone
TPO-Ab Thyroid peroxidase antibody
Ethical considerations
Ethical approval for this case report was waived by the Ethics Committee of Qilu Hospital, Shandong University, as it is a retrospective report of a single patient involving no interventions beyond standard clinical care. The study was conducted in accordance with the Declaration of Helsinki.
Consent to participate
Written informed consent to participate was obtained from the patient.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Author contributions
Linjia Guo, Qiuying Li, Yingqi Liu, Xinyue Yu acquired clinical data, data collection, analysis and interpretation, article writing; Saijia Li performed genetic analysis and interpreted data. Linjia Guo and Qiuying Li wrote the manuscript. Fei Yan critically reviewed and edited the manuscript and approved the submitted version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study received funding from the National Natural Science Foundation of China (NSFC) Regional Innovation and Development Joint Fund (grant number U24A20374), National Natural Science Foundation of China (grant number 81800736), China Endocrine and Metabolic Talent Research Project (grant number 2023-N-03-09), Shandong Provincial Natural Science Foundation (grant number ZR2023MH049), and China Postdoctoral Science Foundation (grant number 2025M782113).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data that support the findings of this study are available from the corresponding author (Fei Yan) upon reasonable request.*