
Abstract
A 70-year-old male with type 2 diabetes presented to the emergency department with severe abdominal pain and dyspnea after 24 weeks of treatment with weekly subcutaneous semaglutide (1 mg). He was diagnosed with acute pancreatitis and diabetic ketoacidosis (DKA) and admitted to the intensive care unit. Following 5 days of stabilization and symptom relief, he was transferred to a general ward. However, the patient subsequently developed a high-grade fever persisting for 3 days, accompanied by severe leukocytosis, despite normalized serum lipase levels. A repeat abdominal computed tomography scan revealed acute acalculous cholecystitis complicated by gallbladder rupture. The patient underwent emergency percutaneous transhepatic gallbladder drainage and antibiotic therapy, leading to gradual recovery. This clinical course illustrates a rare cascade of severe complications in a patient receiving a glucagon-like peptide-1 (GLP-1) receptor agonist. Specifically, when new-onset systemic symptoms emerge following the initial stabilization of a critical illness, clinicians should maintain vigilance for repeating imaging studies. This rare case highlights the need for further investigation into whether the pharmacological effects of GLP-1 receptor agonists, such as biliary stasis, might act as a predisposing factor for complex biliary complications in the setting of severe physiological stress.
Keywords
Introduction
Type 2 diabetes mellitus (T2D) is a global health issue with a rising prevalence, currently affecting an estimated 537 million adults worldwide, with projections reaching 783 million by 2045. 1 The advent of glucagon-like peptide-1 receptor agonists (GLP-1 RAs) has revolutionized the T2D treatment landscape. 2 These agents are widely recognized for superior glycemic control and weight management benefits, with specific agents within the class demonstrating cardiovascular protection, as evidenced in major cardiovascular outcome trials such as SUSTAIN-6 and PIONEER 6.3,4 Consequently, semaglutide, a potent once-weekly subcutaneous GLP-1 RA, has been widely adopted worldwide.
However, with the widespread use of semaglutide and its class, associated adverse events have garnered increasing attention. Beyond common gastrointestinal side effects like nausea, vomiting, and diarrhea, rare but severe complications have emerged. 5 Acute pancreatitis is one of the most well-known potential risks. Although large clinical trials reported a very low incidence without statistical significance compared to placebo, post-marketing surveillance continues to advise clinical vigilance.3,6 Furthermore, the association between GLP-1 RAs and biliary diseases has become a subject of recent research. Meta-analyses suggest that GLP-1 RA use may increase the risk of cholelithiasis and acute cholecystitis, potentially linked to rapid weight loss-induced changes in bile composition.7,8 Notably, most discussions have focused on calculous cholecystitis.
Regarding diabetic ketoacidosis (DKA), it is traditionally not considered a direct side effect of GLP-1 RAs, as their mechanism involves glucose-dependent insulin secretion. 2 While rare cases of DKA have been reported in patients receiving GLP-1 RAs, these events typically occur in the presence of associated risk factors, such as severe beta-cell exhaustion or dehydration.9,10
This case report is unique as it describes a patient who, following semaglutide treatment, developed a rapid succession of three severe, potentially life-threatening complications including DKA, acute pancreatitis, and the far rarer acute acalculous cholecystitis (AAC) complicated by gallbladder rupture. To our knowledge, such a complex constellation of adverse events in a single patient has rarely been reported. We present this case to document its rare clinical course and to explore the potential pathophysiological mechanisms linking these complications, aiming to heighten clinical awareness regarding the possibility of multiple overlapping rare adverse events.
Case report
A 70-year-old male with a 15-year history of T2D and hypertension, presented to our emergency department (ED) with acute onset severe epigastric pain and dyspnea. His active concurrent antidiabetic regimen included metformin 500 mg orally three times daily with meals, 30 units of insulin degludec at bedtime, and weekly subcutaneous semaglutide 1 mg. Semaglutide had been initiated 6 months prior. The weekly dose was up-titrated from 0.25 to 1 mg over approximately 8 weeks, per standard protocol. His most recent dose was administered 5 days prior to ED. Leading up to the acute presentation, the patient reported omitting his metformin and insulin degludec for 3 days due to decreased appetite. The patient reported no routine self-monitoring of blood glucose. His HbA1c level was approximately 14% prior to semaglutide initiation 6 months previously, and was approximately 13% when measured 3 months prior to presentation. He endorsed frequent polyuria and occasional nausea, but declined basal-bolus insulin therapy. On the day of presentation, he experienced sudden, tearing epigastric pain radiating to the back, which was partially relieved by bending forward. Symptoms progressed to shortness of breath within hours. Upon arrival at the ED, the patient was drowsy and slow to respond, though responsive to pain stimuli. He was a retired architect with no history of surgery, drug allergies, or smoking. He reported minimal social alcohol consumption (approximately one standard drink per month) and explicitly denied any alcohol intake in the weeks preceding presentation. His family history is notable for a father with T2D. Furthermore, he had no history of gallstones, cholecystitis, or pancreatitis. The patient is 165 cm tall; his baseline weight before starting semaglutide was 70 kg, and his weight upon admission was 67 kg, reflecting a weight loss of 3 kg over the 6-month period. No recent pretreatment abdominal imaging was available in the cloud-based electronic health record system. Physical examination revealed an elevated blood pressure of 181/74 mmHg, tachycardia (124 bpm), and tachypnea (28 bpm) exhibiting a Kussmaul breathing pattern. Abdominal examination showed significant epigastric and rebound tenderness with hypoactive bowel sounds, though Murphy’s sign was negative.
Arterial blood gas analysis indicated severe metabolic acidosis (pH, 7.159; reference range (RR), 7.35–7.45; HCO3−, 4.6 mEq/L; RR, 22–26 mEq/L) with a significantly elevated anion gap. Biochemistry revealed severe hyperglycemia (632 mg/dL; RR, fasting 7–100 mg/dL), elevated serum ketones (4.3 mmol/L; RR, <0.6 mmol/L), and increased serum osmolality (317 mOsm/kg; RR, 275–295 mOsm/kg). Pancreatic enzymes were markedly elevated (Amylase 374 U/L; RR, 29–103 U/L), Lipase 2013 U/L; RR, 11–82 U/L), exceeding three times the upper limit of normal. Leukocytosis (white blood cell (WBC) count, 8970/μL; RR, 3900–10,600/μL) and acute kidney injury (blood urea nitrogen (BUN), 39 mg/dL; RR, 7–25 mg/dL; creatinine 1.74 mg/dL; RR, 0.6–1.2 mg/dL) were also noted (Table 1). A follow-up report indicated an elevated HbA1c (13.7%, RR, <5.7), mildly elevated triglycerides (171 mg/dL; RR, <150 mg/dL) and a normal serum IgG4 level (48.6 mg/dL; RR, 3–201 mg/dL). An urgent abdominal computed tomography (CT) scan revealed diffuse pancreatic swelling with peripancreatic fat stranding, consistent with acute pancreatitis (Figure 1(a)). The gallbladder was unremarkable at that time with no discernible wall thickening, pericholecystic fluid, or mucosal enhancement (Figure 1(a)). Abdominal ultrasound also showed a normal-sized gallbladder without wall thickening, and no stones or dilation were observed in the biliary system (Figure 2(a)). Based on the clinical presentation, biochemical abnormalities, and CT findings, the patient was diagnosed with DKA complicated by mild interstitial edematous acute pancreatitis, as defined by the Revised Atlanta Classification, and was subsequently admitted to the intensive care unit (ICU). Management included aggressive fluid resuscitation, continuous intravenous insulin infusion, and electrolyte correction. He was kept nil by mouth and treated with analgesics and empiric antibiotics with ceftriaxone. The patient had no history of sodium-glucose cotransporter-2 inhibitor use. DKA (acidemia and ketosis) resolved within the initial 48 h of admission, with concurrent resolution of abdominal pain. Despite this, the patient required intensive care for a total of 5 days for the ongoing management of acute pancreatitis, intravenous fluid resuscitation, and transition to subcutaneous insulin before being transferred to the general ward.
Laboratory findings: comparison between admission and clinical deterioration.
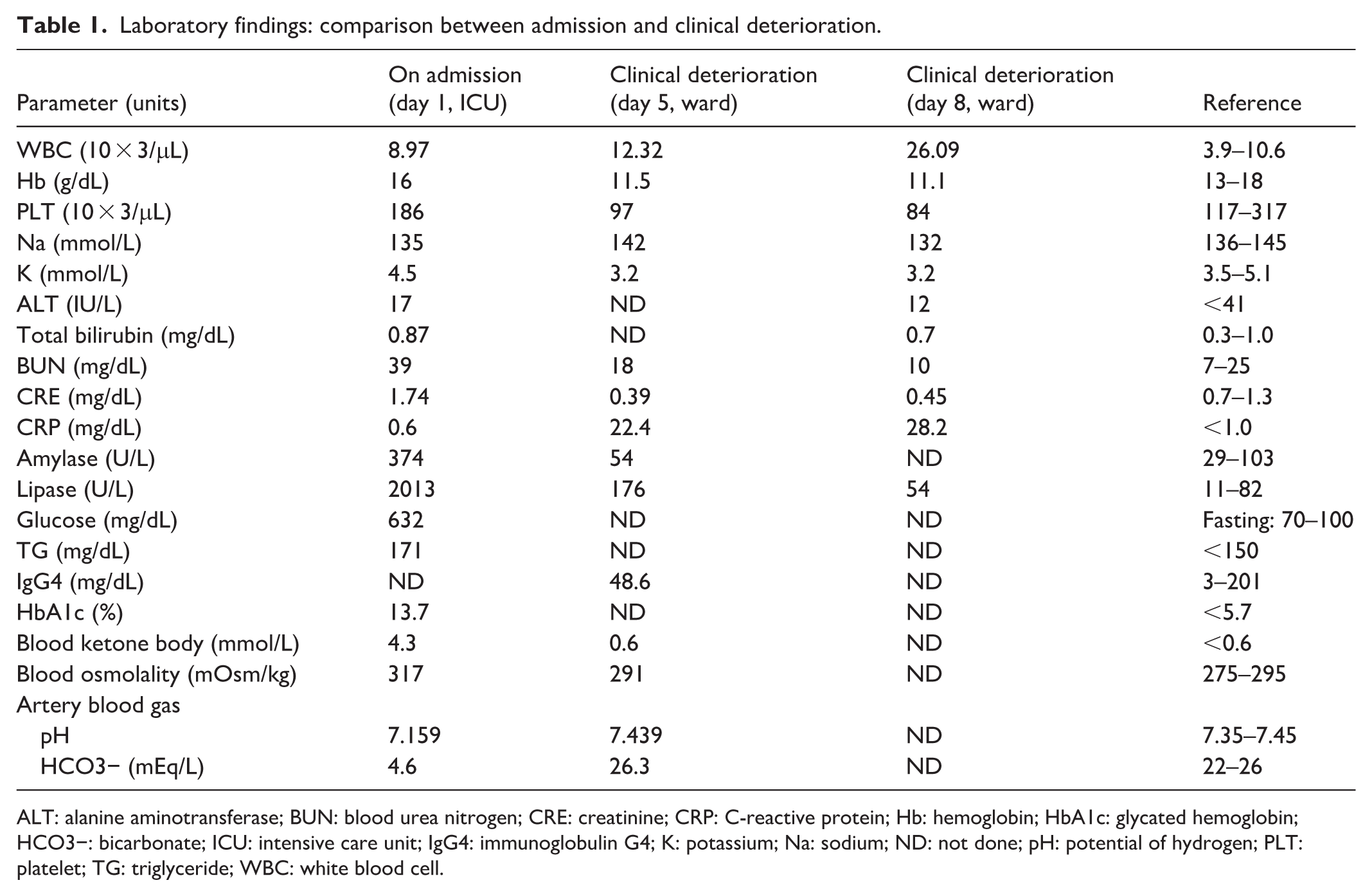
ALT: alanine aminotransferase; BUN: blood urea nitrogen; CRE: creatinine; CRP: C-reactive protein; Hb: hemoglobin; HbA1c: glycated hemoglobin; HCO3−: bicarbonate; ICU: intensive care unit; IgG4: immunoglobulin G4; K: potassium; Na: sodium; ND: not done; pH: potential of hydrogen; PLT: platelet; TG: triglyceride; WBC: white blood cell.
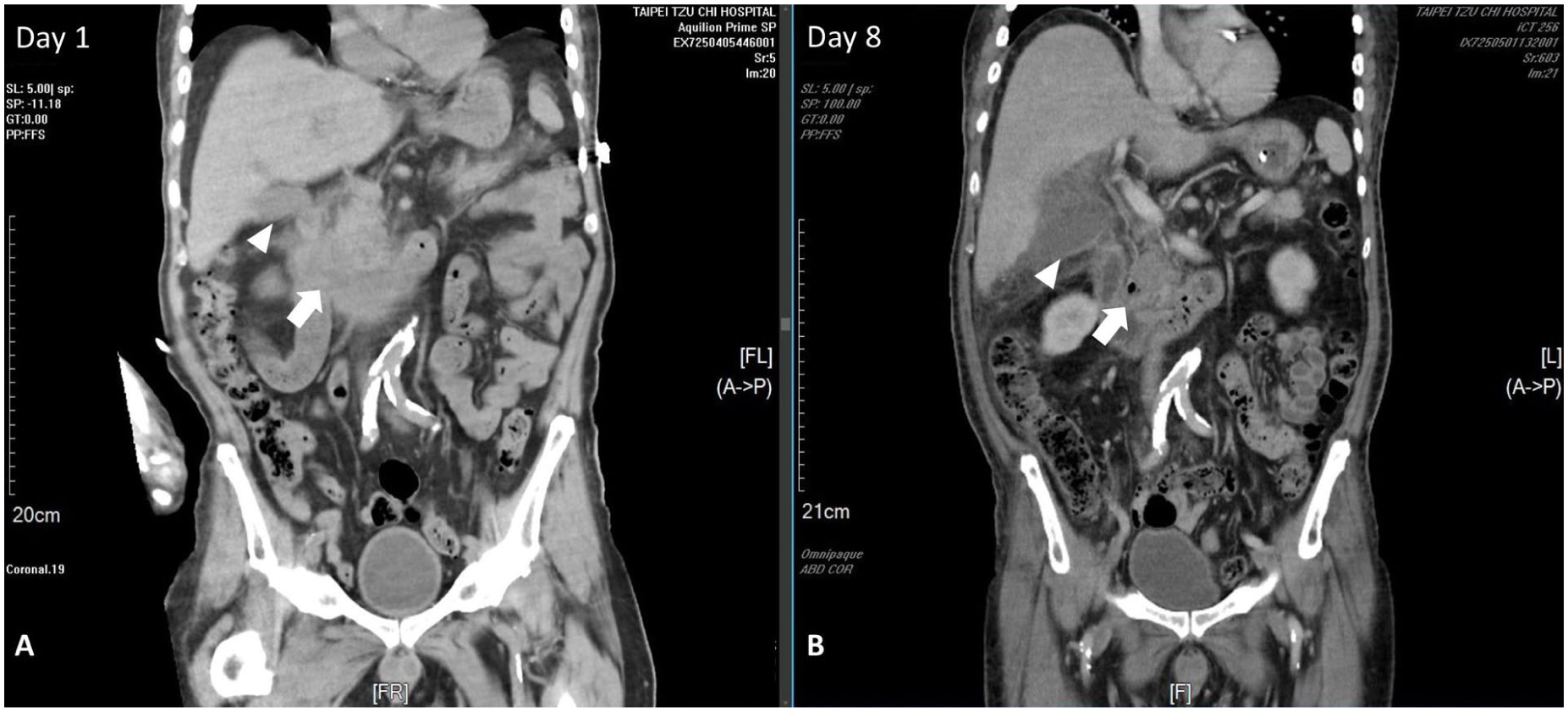
Serial abdominal CT images. (a) Coronal CT image on day 1 demonstrates pancreatic swelling with adjacent fat stranding, compatible with acute pancreatitis (white arrow). The gallbladder appeared unremarkable (white arrowhead). (b) Follow-up CT on day 8 shows significantly improved pancreatic swelling (white arrow); however, findings are highly suspicious for acute cholecystitis with perforation (white arrowhead).
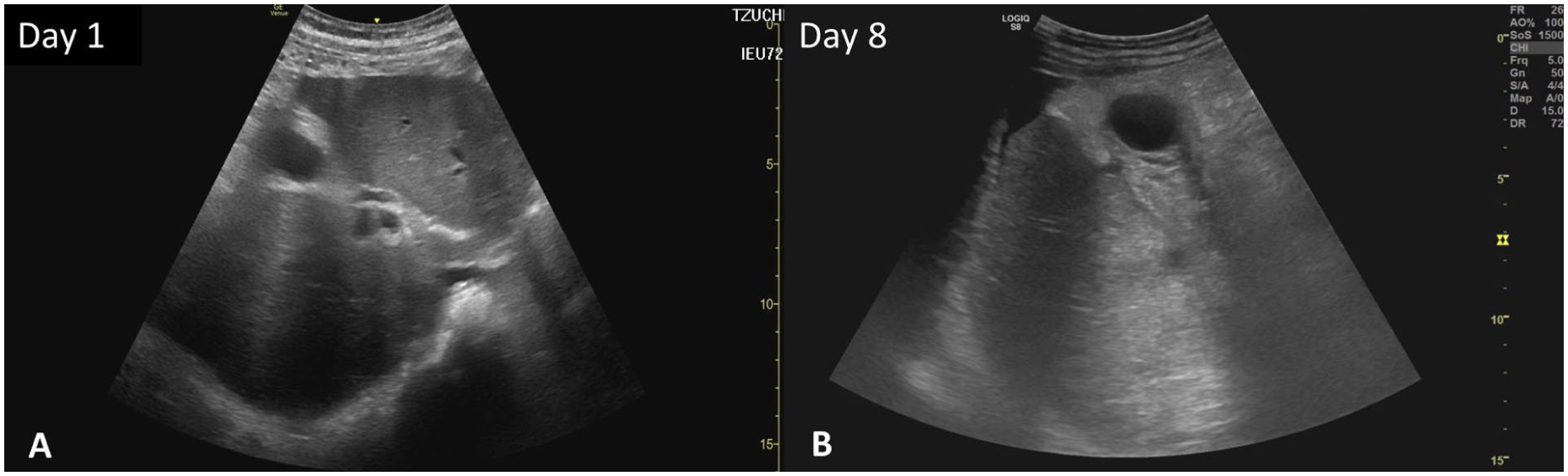
Serial abdominal ultrasound. (a) The abdominal ultrasound on day 1 showed a normal gallbladder without stones or CBD dilatation. (b) The abdominal ultrasound on day 8 showed a cystic lesion near the gallbladder fundus with a small amount of complicated ascites.
However, the clinical course took an unexpected turn on the evening of the transfer. The patient reported recurrent epigastric pain, clinically indistinguishable from his previous pancreatitis pain, accompanied by intermittent fever to 38.3°C. Immediate blood exam revealed an elevated WBC count of 12,320/μL (RR, 3900–10,600/μL) and a CRP level of 22.4 mg/dL (RR, <1.0 mg/dL); conversely, the lipase level decreased to 176 U/L (RR, 11–82 U/L). Consequently, antibiotic therapy was escalated to Brosym (cefoperazone/sulbactam), accompanied by symptomatic treatment and supportive care for suspected sepsis secondary to acute pancreatitis. By day 8, his condition deteriorated rapidly with high-grade fever (39.1°C), chills, and intensifying right upper quadrant (RUQ) pain. Physical examination revealed a lethargic, diaphoretic patient with signs of severe systemic inflammatory response, tachycardia, hypotension, and signs of localized peritonitis in the RUQ. Laboratory tests showed a surge in WBC to 26,000/μL (RR, 3900–10,600/μL) with a left shift, while pancreatic enzymes remained stable (Table 1). A second urgent abdominal CT proved to be key to the diagnosis, presenting a striking contrast to the admission scan taken 8 days earlier. The original pancreatic inflammation had subsided; however, the gallbladder appeared severely distended with a significantly thickened wall, showing clear signs of submucosal edema and surrounding fat stranding. Crucially, the scan showed a focal defect in the gallbladder wall with a contained pericholecystic fluid collection, highly suggestive of gallbladder rupture (Figure 1(b)). The biliary system remained free of stones or ductal dilatation. Follow-up abdominal ultrasound revealed a cystic lesion at the gallbladder fundus, accompanied by a small amount of complex ascites (Figure 2(b)).
The final diagnosis was sepsis secondary to AAC with gallbladder rupture. Antibiotics were escalated to doripenem. Given a concern of his recent recovery from DKA and acute pancreatitis with severe catabolic state, the surgeon strongly recommended emergent percutaneous transhepatic gallbladder drainage (PTGBD) as the safest immediate intervention. The procedure drained 80 mL of thick, purulent bile, confirming severe infection. Following drainage, sepsis resolved rapidly; fever subsided the next day, and leukocyte counts normalized shortly thereafter. Blood cultures were drawn concurrently with the onset of the severe leukocytosis and high-grade fever; however, they all were negative. The bile culture obtained via PTGBD grew Escherichia coli. Susceptibility testing revealed resistance to ceftriaxone (minimum inhibitory concentration (MIC) >64 µg/mL) but susceptibility to doripenem, justifying our clinical escalation of antimicrobial therapy. The patient was discharged after 2 weeks of antibiotics and PTGBD. Six weeks later, the surgeon recommended cholecystectomy; however, the patient expressed relief following the drainage procedure but opted for conservative management due to personal concerns regarding surgical risks. After removal of PTGBD, he remains free of recurrence over 8 months of follow-up, and his blood glucose remained controlled with insulin-based treatment (Figure 3).
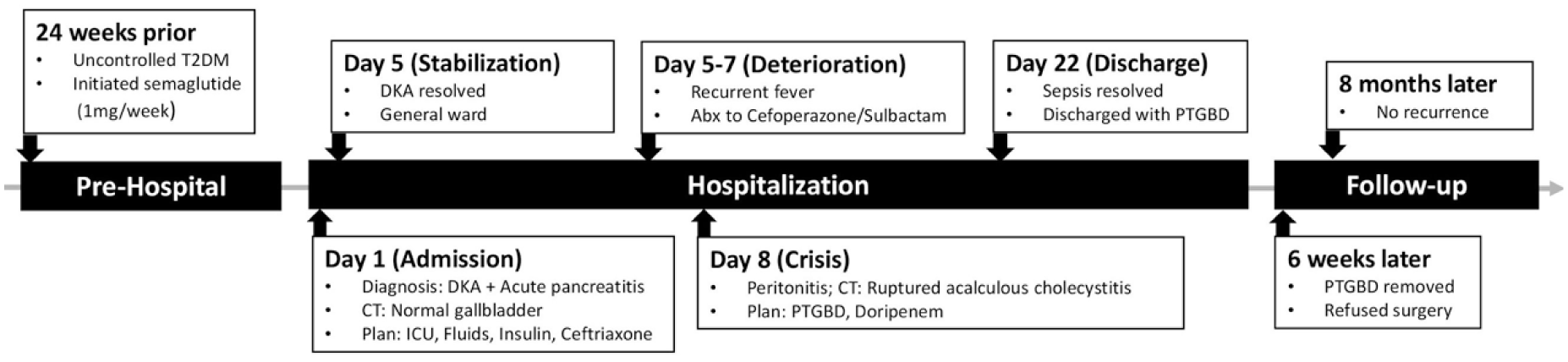
Timeline of the clinical course. The figure illustrates the sequence of events from the initiation of semaglutide to the development of DKA and acute pancreatitis, followed by the delayed onset of ruptured acalculous cholecystitis.
Discussion
This case reports a T2D patient with poor glycemic control treated with semaglutide who developed sequential DKA and acute pancreatitis, followed rapidly by life-threatening AAC with gallbladder rupture. To clarify the acalculous nature of the cholecystitis, the initial day 1 CT scan was reviewed with thin-slice reconstructions, showing no radiopaque stones, sludge, or biliary dilatation. Additionally, a day 8 abdominal ultrasound, a highly sensitive modality for sludge and microlithiasis, confirmed the absence of echogenic material, stones, and common bile duct dilatation, solidifying the diagnosis of true AAC. To our knowledge, such a complete and severe progression in a single patient is exceptionally rare, warranting a discussion on the underlying pathophysiology.
First, the role of semaglutide in this patient’s DKA must be clarified. GLP-1 RAs stimulate insulin secretion in a glucose-dependent manner and are not typically considered a direct cause of DKA. 2 While the lack of prior continuous glucose monitoring precluded the assessment of time in range, the patient’s HbA1c of 13.7% alone strongly suggests near-total beta-cell exhaustion, mimicking a state of absolute insulin deficiency. 11 Although a serum C-peptide level was unfortunately not obtained during this acute admission to definitively confirm this, which represents a limitation of this retrospective report, his 15-year history of T2D strongly supports profound beta-cell dysfunction. However, this severe clinical presentation was likely multifactorial; the profound hyperglycemia and elevated HbA1c could also have been compounded by other factors, such as medication non-adherence or the acute physiological stress of his developing illness. In such patients with minimal insulin reserve, any physiologic stress including infection or dehydration, can rapidly precipitate metabolic decompensation. The gastrointestinal side effects of semaglutide may have led to reduced intake and dehydration, triggering ketogenesis.9,12 Thus, while semaglutide was likely not the direct etiology, its potential side effects may have served as a precipitating factor in a patient with severely compromised islet function.
Second, regarding acute pancreatitis, the link to GLP-1 RAs remains debated. While trials like SUSTAIN-6 showed no significant risk increase, post-marketing reports persist.3,6 In this case, DKA itself is a known risk factor for acute pancreatitis; severe acidosis, dehydration, and hypoperfusion can damage the pancreas. 13 Furthermore, the absence of hypertriglyceridemia and normal IgG4 levels preclude these conditions as underlying etiologies. Consequently, determining whether the acute pancreatitis was a complication of DKA, a rare drug side effect, a precipitating factor for DKA, or simply idiopathic remains challenging, as these conditions are likely interrelated.
The most critical turning point in this case was the development of AAC and subsequent gallbladder rupture following the initial stabilization of DKA and acute pancreatitis. This clinical presentation differs fundamentally from the typical calculous cholecystitis frequently associated with GLP-1 RA-induced weight loss. We acknowledge that AAC is a recognized complication in critically ill patients, primarily driven by systemic hypoperfusion and ischemia. However, typical illness-induced AAC develops insidiously over 1–3 weeks of intensive care, with frank gallbladder rupture occurring in only 10%–15% of cases.14 –16 In stark contrast, our patient experienced a remarkably rapid progression to gangrene and perforation. This atypically accelerated clinical course strongly suggests the presence of an additional, synergistic insult. We propose a multi-hit cascade mechanism to explain this catastrophic deterioration. The first hit was the systemic stress originating from DKA and acute pancreatitis, which induced systemic inflammatory response syndrome and hypovolemia. To maintain perfusion to vital organs, splanchnic vasoconstriction occurred, resulting in profound gallbladder wall ischemia.14,15 The second hit involved severe gallbladder stasis. While standard critical care measures, such as prolonged fasting, contribute to physiological stasis by halting cholecystokinin secretion, semaglutide exerts a distinct and potent inhibitory effect on gallbladder motility and gastric emptying. We posit that this persistent medication-induced dysmotility acted as a critical catalyst, severely amplifying the physiological stasis. 17 While evidence demonstrates that liraglutide significantly inhibits gallbladder motility, 17 direct data regarding semaglutide’s specific effect on biliary stasis is limited. Extrapolating these findings to semaglutide is speculative; however, given the established class effect of GLP-1 RAs on delaying gastric emptying, a concurrent impact on gallbladder dynamics remains a plausible, albeit unconfirmed, contributing mechanism.
The synergistic convergence of illness-induced ischemia and pharmacologically exacerbated stasis precipitated rapidly progressive AAC. Consequently, the highly ischemic and gangrenous gallbladder wall became exceptionally susceptible to secondary bacterial translocation from the gut—manifesting clinically as sudden leukocytosis—which ultimately culminated in frank rupture and subsequent E. coli sepsis. 18 This synergistic mechanism elucidates why the catastrophic rupture occurred sequentially and with such unusual rapidity following the initial acute event. The definitive treatment for AAC with gallbladder rupture is cholecystectomy. Conservative management with PTGBD as a standalone therapy is generally reserved for patients unfit for surgery, as it carries a substantial risk of recurrent biliary sepsis, gallstone formation, and chronic cholecystitis, particularly in diabetic patients who are inherently vulnerable to severe infections. Our patient’s decision to refuse surgery represents a deviation from standard guidelines, necessitating close, long-term clinical surveillance. Crucially, as this is a single case report, a definitive causal role of semaglutide cannot be confirmed. The profound systemic stress and ischemia from the preceding DKA and pancreatitis were the primary drivers, while semaglutide likely acted as a predisposing factor by contributing to underlying biliary stasis.
Conclusions
The clinical significance of this case report lies in highlighting a potentially hazardous clinical course in a patient receiving semaglutide. First, in patients with markedly elevated HbA1c, which may indicate near-exhaustion of pancreatic beta-cell function, the use of GLP-1 RAs requires caution, as their associated gastrointestinal adverse effects could potentially contribute to or exacerbate the risk of DKA. Second, this case illustrates that biliary complications in patients receiving GLP-1 RAs are not exclusively calculous in nature. The pharmacological effect of biliary stasis may act as a critical predisposing factor for AAC when compounded by the severe ischemia of critical illnesses. Therefore, in patients recovering from severe conditions such as DKA or acute pancreatitis, the onset of new fever or abdominal pain necessitates the inclusion of AAC as a critical differential diagnosis. Finally, repeat abdominal imaging within a short interval should be promptly considered to prevent catastrophic outcomes.
Footnotes
Acknowledgements
The authors utilized AI assistance (Gemini Pro 3.0, Google) for the translation and linguistic refinement of the manuscript. The final content was meticulously reviewed and verified by the authors to ensure clinical accuracy, and the authors assume full responsibility for the final submission.
Ethical considerations
The case report has been approved for publication by the Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation
Institutional Review Board (No. 14-IRB149).
Consent to participate
Written informed consent for publication was obtained from the patient.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed during this study are included in this published article.