
Abstract
All new drugs targeting or influencing the central nervous system (CNS) must be screened for Drug Abuse Liability (DAL) prior to license approval by the FDA. Drug discrimination, self-administration, and drug dependence potential study designs are three core behavioral assays proposed in the 2017 FDA Guidance to Industry on Abuse Liability Testing for submission to the agencies for review at the time of the NDA submission. There are no international or federal drug control agency requirements for which animal species to use and selection of the test parameters for the sex, strain, age, dose range, study duration, systemic drug exposure thresholds or positive comparators to use in the conduct of these studies. In pre-IND and pre-NDA discussions with sponsor representatives, it is the FDA that has placed the financial burden on the industry to conduct these studies in both sexes in, what appears to be, a direct conflict with the intent of the Animal Welfare Act (1996). There is no single drug-of-abuse that is self-administered exclusively by one sex and there are no differential schedule controls placed on any drug substance based on any sex- or gender-based pharmacokinetic parameter. These nonclinical assays used for drug control scheduling actions should be conducted in only one sex unless there is a strong indication that sex is an important factor in the therapeutic use of the new drug or the mechanism of action.
Keywords
Introduction
The preponderance of available epidemiologic data on drug abuse patterns in the U.S. show that males consistently enter the health care system for medical treatment related to the adverse effects of drug abuse (detoxification, medical treatment, or psychological services) more often than their female contemporaries. McHugh et al. (2010, 2017a, 2017b) recently summarized the current ambiguity between sex and gender as it relates to substance use disorders (SUDs). There is a greater prevalence of SUDs in men. While biological sex differences are evident across an array of systems, including brain structure and function, endocrine function, and metabolic function McHugh et al. (2010, 2017a, 2017b) concluded that gender differences contribute to the initiation and course of SUDs It is evident that men and women do not substantively differ with respect to SUD treatment outcomes. McHugh et al. (2017b, 2018) conclude that several trends are emerging in the literature to suggest that sex differences in SUDs are complicated by the interactive contributions of biological and environmental factors.
As recently summarized by González-Rojano et al. (2018, 2019): The European Medicines Agency’s (EMA) guideline on the investigation of bioequivalence allows the inclusion of only men in studies conducted with drug products that will be administered also to women, because the subjects recruited for bioequivalence studies can belong to either sex (EMA, 2010). Even in the case of products that will be used only for women, the inclusion of only males is acceptable and sometimes recommended, e.g., to avoid endogenous levels of hormones if the exogenous administration of those hormones is under comparison. In addition, this guideline indicates that the risk to women of child bearing potential should be considered, which is another reason to avoid the inclusion of women. In contrast, the United States Food and Drug Administration (FDA) guideline recommends that if the drug product is intended for use in both sexes, similar proportions of males and females should be recruited in the bioequivalence study. (United States Food and Drug Administration, 2003)
The NIH and FDA have adopted full adherence to the U.S. Animal Welfare Act (https://grants.nih.gov/grants/guide/notice-files/not93-071.html) which requires all research institutes limit animal use in research. However, in 1994 the NIH established guidelines on the inclusion of women and minorities and their subpopulations in research involving human subjects, including clinical trials that were supported by the NIH Revitalization Act of 1993 (Federal Register, March 9, 1994 [59 FR 11146-11151], https://grants.nih.gov/grants/guide/notice-files/not94-100.html.) Originally, this action was not binding on nonclinical research or the pharmaceutical industry. In 1998 the FDA amended its regulations pertaining to new drug applications (NDAs) to clearly define the requirement to present effectiveness and safety data for important demographic subgroups; specifically, gender, age, and race (Federal Register, February 11, 1998 [63 FR 6854-6862], (https://www.fda.gov/ScienceResearch/SpecialTopics/RunningClinicalTrials/ucm120254.htm) . The final rule did not increase the requirements for the clinical study conduct nor did it require sponsors to conduct additional studies or collect additional data. The 1998 FDA final rule referred only to the presentation of data already collected. Faulty assumptions stemming from this under-representation of females in research has been dispelled by a rapidly increasing body of published reports appearing in peer-reviewed scientific journals (Epstein and Menges, 2013; Greenfield et al., 2007, 2010a, 2010b, 2011).
It was the original intent of the NIH that sex as a biological variable (SABV) would be factored into research designs, analyses, and reporting in vertebrate animal and human studies submitted to
NIH policy stated the importance of determining whether the preclinical pharmacotherapy studies and other translational studies would apply to both sexes, while noting that some sex differences that emerge may not be meaningful or interesting. For example, sex-dependent body size differences could affect activity in behavioral testing apparatuses without influencing the main variables of interest. While pharmacokinetic differences (Cmax and AUCs) between males and females have been identified to be the result of body weight differences (mass or volume) these have not been the basis of regulatory decision-making determinations on schedule control status.
The CSS of CDER at FDA may not accept abuse liability data gathered from a single test system (even if it is qualified) for an investigational drug unless FDA concludes there is sufficient evidence that the results generated in the species adequately predicts the response to the drug once licensed for human use. The regulatory decision to allow approval of a drug based on the use of the three animal models to predict human abuse potential in rats will be made by CSS on a case-by-case basis at the time of the NDA review. Since the DAL animal studies are not typically conducted until the end of Phase II clinical trials, when the final therapeutic doses are usually selected, the decision to accept or reject the DAL data will not be determined until the latter stages of Phase III human clinical trials. Major decisions this late in the drug approval process may represent a logistics nightmare for the drug development team and financial officers of the company.
As a Contract Research Organization (CRO) that routinely conducts abuse liability studies for drug development companies, we always encourage each sponsor to submit protocols to the Controlled Substances Staff (CSS) of the Center for Drug Evaluation and Research (CDER) at FDA for review and input prior to the initiation of the fully Good Laboratory Practice-compliant (GLP) study protocols. Using 4 recent “drug abuse liability screening programs” here at CRL-Mattawan as examples, we received rapid, cogent, and detailed feedback from the CSS on the sponsor-submitted protocol packages (3 study protocols per package) that included variants of using both sexes and single sex study designs. The proprietary details of these privileged communications are contractually limited, however with respect to the use of animal subjects the CSS responses in all cases was: Males and females should be used.
It is interesting to note that justification for the use of only one sex was provided in all submitted protocols with demonstrative evidence of non-significant differential sex-dependent PK and PD findings in other nonclinical IND-enabling studies conducted with the compounds.
Under DAL Guidelines (FDA, 2017) the current thinking of the FDA is that while it is appropriate to use animals of both sexes in these studies (Section IV[D][1], page 15) the agency has approved the last 5 new schedule controlled drugs based on abuse liability tests conducted in only one sex of laboratory animal: Lorcaserin (Belviq®), Suvorexant (Belsomra®), Eluxadoline (Viberza®), Rolapitant (Varubi®) and Briveracetam (Briviact®), (see Table 1).
The five most recently FDA approved NDAs and the subsequent schedule controlled actions taken by the DEA.

All five of the most recently approved schedule-controlled drugs had sexed-based differential PK data showing higher Cmax and AUCs for females than males, yet no differential labelling, dosage regimens, or schedule control recommendations were proposed by FDA. Based on these data alone, there is questionable value in the evaluation of both sexes in DAL assays.
Similar to the Animal Rule, under the current set of International Council on Harmonization (ICH) guidelines adopted by the FDA, not all nonclinical studies require both male and female animal subjects, the decision of which should be conducted as is appropriate for the population that is being targeted. Part of every licensure application of new drugs that act directly or indirectly on the central nervous system (CNS) must include a series of nonclinical studies that have predictive validity for abuse liability in the clinical population. (FDA, 2017). The current disparity in sex-dependent differences in both drug abuse patterns and treatment seekers in the US appears to support the minimal use of male subjects in the NDA-enabling abuse liability testing scheme. Since:
there are no known drugs of abuse that are used exclusively by either male or female patients; the preponderance of all abuse liability studies published in peer-reviewed scientific journals report the exclusive use of only one sex (mostly males); international and national laws regulate and place controls on drug substances based on the pharmacology of the test article and do not have statutory discretion to establish legitimate bifurcated schedules of control based on any sex-based pharmacokinetic differences; administrative precedence has been established by the FDA and DEA in establishing drug control policies based solely on single-sex nonclinical abuse liability data sets; And the Controlled Substances Act establishes 8 factors determinative of schedule control actions on new chemical entities that have been approved as medicines in the US by the FDA. There is not a single use of the term “pharmacokinetics” or “sex-based” differences in the CSA. Regardless of a significant difference in drug kinetics between male and females, there is no administrative remedy that can establish differential schedule controls based on sex- or gender-based abuse liabilities.
Based on these five principles, the Animal Welfare Act appears to supersede the administrative policies of the FDA in this matter. There is no legal requirement or legislative history that requires these 3 DAL studies to be conducted with both sexes. Administrative regulations of the FDA are not legally binding on the agency nor the pharmaceutical industry with respect to these studies. In fact, the 2017 FDA guidance document clearly states: This guidance represents the current thinking of the Food and Drug Administration (FDA or Agency) on this topic. It does not establish any rights for any person and is not binding on FDA or the public. You can use an alternative approach if it satisfies the requirements of the applicable statutes and regulations.
We submit the legal premise that the 8 factors determinative of schedule control recommendations by the FDA are fulfilled by data collected in only male rats. The preponderance of all available nonclinical data from self-administration, drug discrimination, and dependence liability studies support the statutory requirements of the Animal Welfare Act (1996) and the International Council on Harmonization Guidelines (M3 [R2], 2000) to reduce the tests to only one sex (males) in the conduct of all nonclinical abuse potential studies. Further, the extensive resources required for conducting DAL testing of all new drugs in both sexes is incongruent with the recent administrative “least burdensome” policies of the FDA that have been applied to other types (e.g., medical device) approvals (FDA, 2019): We define “least burdensome” to be the minimum amount of information necessary to adequately address a relevant regulatory question or issue through the most efficient manner at the right time. This least burdensome definition considers the type of information, different approaches to generating or providing information, and when during the total product lifecycle information should be generated or provided to FDA.
Under the “least burdensome” principles, the FDA seems to allow alternative approaches, when appropriate, to optimize time and resources. Further, FDA acknowledges that there are often multiple ways to satisfactorily address a regulatory issues. The resolution of the regulatory issue should be based on a discussion about which method is least burdensome, while still satisfactorily addressing the regulatory issue. We propose that such policies include the use of only one sex in DAL testing.
Additionally, the FDA has also adopted regulatory guidelines that delineate study designs for the preclinical screening of food ingredients (United States Food and Drug Administration, 2007). FDA requires that only some animal studies required for approval include both males and females. Current safety pharmacology core battery studies (cardiovascular, pulmonary and CNS) are generally conducted in only one sex. Clinical and experimental studies show that females clearly demonstrate sex-dependent differences in the electrocardiographic pattern of ventricular repolarization in animal species and in humans. These electrocardiographic (ECG) changes are associated with a longer rate-corrected QT (QTc) interval at baseline than males (Abi-Gerges, 2004). While there is limited documentation regarding drug differences between women and men, data from drug adverse events have shown that women experience torsades de pointes, a potentially fatal arrhythmia, more frequently than male cohorts (Anthony, 2005). Despite these discrepancies, the majority of all cardiovascular safety studies are conducted using males (Gauvin et al., 2009).
Other second tier assays described in ICH S7A and M3(R2), such as renal, gastrointestinal (motility and stomach emptying), and neurological assays (EEG, seizure sensitivity) are generally conducted in only one sex, as well. For example, since male albino rats generally show proteinuria, Tier II renal studies are generally conducted in female rats, while GI and EEGs are generally only conducted in males.
Drug abuse liability assessment is only one aspect required for NDA approval. Drug control policy is sex-neutral. If there is diversion of a drug product in clinical trials by males or females, or positive preclinical behavioral data that have been shown to have moderate to strong predictive validity for human abuse potential, such as increases in locomotor activity or self-administration in either male or female rats, then drug scheduling reviews are initiated: the FDA/CDER guidance provides a considerable degree of scientific flexibility regarding models and methodology, because the assessment of abuse potential of a drug is an evolving area of research…There is no mention in the CSA of the type of medical and scientific data required to assess abuse potential for scheduling purposes. (Calderon and Klein, 2014)
The critical question
DAL testing is required for all new CNS-active drugs under the Controlled Substances Act (1970). Under the CSA, the 8 factors determinative of schedule control actions are blind to sex differences. The CSA does not give more regulatory weight to any contributing factors related to a drug’s pharmacokinetics, such as Cmax and AUCs. The purpose of this review is to establish a rationale for standardized preclinical protocols related to the 3 core behavioral assays used to assess a new drug’s abuse liability that must be submitted to support NDA approval: 1) drug discrimination, 2) self-administration, and 3) dependence potential assessments.
Pharmacokinetics: Drug control and sex
Human bodies have developed a means to rapidly metabolize and eliminate thousands of structurally diverse foreign chemicals which has been critical in adapting to changing environments. Drug metabolizing enzymes have evolved and are involved in detoxifying a multitude of endogenous chemicals as well as those consumed as medicaments, intoxicants, and food supplements. Most of these xenobiotics are lipophilic chemicals, and in the absence of metabolic enzymes these substances would simply accumulate in the body to the point of toxicity. With a few exceptions these xenobiotics are subjected to enzymatic pathways that constitute a first phase oxidation, reduction or hydrolytic reactions and a second stage conjugation process by which enzymes catalyze the products of phase I and conjugate that with a second molecule (Gonzalez et al., 2018). Phase I oxidation reactions are carried out by cytochochrome P450 superfamily of isoenzymes (P450). P450 2D6 isoenzyme has been implicated in at least 30 different drug oxidation reactions (Phase I) and has been reported to have considerable racial differences in its expression and function. The P450 3 A subfamily of enzymes play a more prominent role in human metabolism of xenobiotics (Guengerich, 1995). Wolbold et al. (2003) reported a substantial degree of evidence to support a role of sex in the control of CYP3A4. Many drugs metabolized by CYP3A4 show higher clearance in women compared with men even with body weight corrections for Cmax and AUCs. Commonly abused drugs metabolized through CYP3A4 include anabolic steroids, opiates, and benzodiazepines. However, quantitative differences in CYP3A4 expression between men and women are not convincingly established despite a number of studies failing to show any differences (George et al., 1995; Schmucker et al., 1990; Shimada et al., 1994; Transon et al., 1996). In 2003, Wolbold et al. provided convincing evidence that CYP3A4 is differentially expressed in men and women, with women expressing about twice the amount of the enzyme on the basis of microsomal protein content. These data explain why women tend to clear some CYP3A4 substrates faster than men but there are exceptions to the rule (for example, midazolam).
The physical properties of the drug preparation, boundary characteristics (membranes) separating the site of drug entry and action will affect the onset of drug action and the time of peak effects. For example, it is generally presumed that direct injection into a muscle provides faster onset of action relative to oral administration, but this is not always the case. One of the first water-soluble anti-anxiety medications used in the treatment of alcohol detoxification, Librium® (chlordiazepoxide) is usually prescribed for oral administration. Consuming a tablet of Librium® produces rapid and complete absorption, with subjective patient reports of feeling “spacey” within 15 minutes (Greenblatt et al., 1974, 1977a, 1977b). Intramuscular injection of Librium® produces relatively slow, erratic and incomplete absorption due to its lipophilicity, causing the drug to be deposited in systemic fat and not into the bloodstream for transport into the brain (Greenblatt, et al., 1974, 1978; Robinson et al., 1983). Regardless of the differential onset of effects of Librium®, the drug substance was placed in Schedule IV of the CSA.
The most critical factor to keep in mind in this discussion of sex-based differences in abuse liability data is that drug control policy decisions are based on drug substances, not drug formulations. Sex-based differential absorption, distribution, metabolism and excretion (ADME) of formulated drug delivery systems are not determinative factors in assessing relative abuse liability under the CSA. For example, the differential routes-of-administration and pharmacokinetic profiles of fentanyl provided by slow release Duragesic® (dermal patch), immediate release formulations like Abstral® (sublingual tablet), Actiq® (oral lozenges), Fentora® (buccal tablet), Lazanda® (nasal spray), and Subsys® (sublingual spray), or the intravenous formulation, Sublimase® do not support a differential scheduling of these products; all FDA-approved formulations containing the drug substance, fentanyl, are in Schedule II of the CSA. Differential onset of action (Tmax) is not a determinative factor in placement of new drugs into differential schedules of control. As history has already demonstrated, the controlled release matrix of OxyContin® and the sex-based differential kinetic profiles of the drug did not diminish or impede the popularity and diversion of the opiate analgesic following license approval in 1996.
González-Rojano et al. (2018, 2019) have recently published a review of bioequivalence data to investigate the existence of a sex-by-formulation interaction in all bioequivalence studies of transdermal patches submitted to the Spanish Agency for Medicines between 2010 and 2016. Five different transdermal formulations were reviewed, one buprenorphine patch, 2 variations of fentanyl patch, and 2 variations of rivastigmine patches. The opiate-based buprenorphine and fentanyl patches were approved for pain control and their metabolism by hepatic CYP450 2D6 and 3A4 enzymes show female: male differential kinetics. But rivastigmine is a reversible acetylcholinesterase inhibitor for the treatment of mild to moderate dementia caused by Alzheimer’s or Parkinson’s diseases and is extensively metabolized primarily via cholinesterase-mediated hydrolysis and then renal excretion of the metabolites. Aside from the differential metabolic pathways of the drug, González-Rojano et al. (2019) highlighted the similarity in these approved transdermal drugs based on the fact that they all exhibit three common properties: low molecular weight (MW < 500 Da), balanced lipophilicity (log p = 1–3) and measurable solubility both in oil and in water since the drug has to cross the lipophilic stratum corneum and be absorbed into the systemic circulation. The authors of this review found some minor sex-by-formulation interactions were detected but only in pharmacokinetic parameters derived from some small sampled studies. When replicated in larger populations these differences were excluded. González-Rojana et al. (2019) reported that the sex-based differential bioequivalence data are not reproducible and therefore, at the present time, there is no convincing evidence to require bioequivalence demonstration for transdermal patches in males and females, separately We argue these conclusions generalize to nonclinical abuse liability testing.
Schwartz (2003) and Meibohm et al. (2002) have pointed to the fact that on average, men are larger than women. Body size differences result in larger distribution volumes and faster total clearance of most medications in men compared to women and body fat may increase distribution volumes for lipophilic drugs in females. When all factors are reviewed, total drug absorption is not significantly affected by sex despite slightly slower absorption rates in females. Bioavailability following oral dosing, for CYP3A substrates in particular, may be somewhat higher in females than males. Renal processes of glomerular filtration, tubular secretion, and tubular reabsorption appear to be faster in males than females (on a mg/kg or total body weight basis). While the animal-to-human PK data link is useful and sometimes very important, it is seldom essential during a drug development program (Welling, 1995) as is highlighted by the following selected examples:
Hydrocodone (Vicodin)
The unique interplay between these Phase I and Phase II metabolizing processes and the significance of pharmacokinetics on drug control policy may be best characterized by the most prevalently prescribed and abused opiate analgesics in the US, hydrocodone (i.e., Vicodin). Vicodin is most exclusively prescribed and used recreationally by the oral route of administration. Hydrocodone is primarily metabolized by CYP 2D6 to the O-demethylated active metabolite, hydromorphone, and by the Phase II CYP3A4 enzyme to form the N-demethylated product, norhydrocodone (Hutchinson et al., 2004). The affinity of hydromorphone at the µ-opioid receptor is 30-fold higher than hydrocodone, itself. Therefore, the metabolism of Vicodin influences its analgesic efficacy. In a recent retrospective study by Barakat et al. (2014) gender and other factors are reviewed (age, urinary pH and concurrent use of CYP2D6 and CYP3A4 inhibitors) that contribute to the metabolism of the parent hydrocodone molecule to its urinary metabolites (hydromorphone and norhydrocodone). Barakat et al. reported that the inactive norhydrocodone mole fractions were higher in women, while the more potent metabolite, hydromorphone, (mole fractions) were higher in men. These findings are consistent with the well-established higher CYP3A4 activity levels in female liver microsomes. These data suggest that men metabolize hydrocodone to the more potent hydromorphone “active metabolite” when compared to women.
Table 2 shows the differential emergency department episodes for men and women over an 8-year collection period. Drug abuse and adverse events were more prevalent in women than men for all 8 years of data in Table 1. Despite the different sex-dependent metabolism of hydrocodone in men and women, Cicero et al. (2013) have reported that Vicodin is preferred as first choice medication in women.
Estimated emergency department visits related to nonmedical use of hydrocodone combination products (e.g. Vicodin): 2004 to 2011 DAWN data.

Zolpidem
Another example of the major pharmacokinetic issues regarding nonclinical data reviewed for NDA approval is the sedative/hypnotic, zolpidem. Zolpidem was approved for use in the United States in April 1992, under NDA #19-908 (Ambien™). In the NDA overview dated November 191,991 a number of indices of abuse potential were reported in clinical trials and cited in the pharmacological review such as amnesia, euphoria, depression, and hallucinations. On page 54 of the NDA review document, FDA states, “We did not take zolpidem to the PDAC” meaning the Psychopharmacological Drugs Advisory Committee Meeting. On page 79 of the same review, FDA summarized the “variations in pharmacokinetics” known at the time of NDA approval: Females given zolpidem had a Cmax and AUC approximately 25% higher than males when corrected for weight.
Under general circumstances the abuse liability studies would be conducted in the most sensitive sex – females; however all nonclinical and clinical DAL studies were conducted in only males. Zolpidem was approved and placed into Schedule IV despite the higher PK profiles in females.
For each of the 8 years of the DAWN data (2004–2011), women entered the emergency rooms related to a self-reported consumption of zolpidem more than males. Questions arise regarding the role of the known pharmacokinetic differences (i.e., 25% higher Cmax and AUC) in these adverse event statistics. If the least sensitive sex (males) demonstrated positive preclinical biomarkers for abuse liability, it would be generally assumed, based on the preponderance of similar reports in peer-reviewed scientific journals, that females would show similar biomarkers, albeit possibly at lower doses.
As listed in Table 3, emergency department admissions for zolpidem abuse in females has exceeded those of male patients for the 8 years listed in the publicly summarized data by SAMHSA. These facts may be interesting for labelling purposes during the NDA review, but not for drug control policy decisions. Drug control policy is sex-blind. These facts are irrelevant to drug control policy – male or female – the substance remains correctly controlled with other benzodiazepine sedative/hypnotics.
Estimated emergency department visits related to nonmedical use of Zolpidem (Ambien®): 2004 to 2011 DAWN data.

Buprenorphine
Moody et al. (2011) reported a retrospective study to address sex differences in buprenorphine treatment. Moody et al. reported statistically significant sex differences in pharmacokinetics of buprenorphine in men and women in treatment. The AUCs for plasma buprenorphine concentrations were approximately 39% higher in females (58.4 ± 24.8) compared to males (41.9 ± 14.5; p < 0.01) and an approximate 35% higher Cmax in females (6.97 ± 2.98) compared to males (5.15 ± 1.94; p < 0.05). There were no differences in the time to peak effect (Tmax). With equivalent doses of buprenorphine, the females in the Moody et al. (2011) study were found to have higher plasma concentrations of the drug, yet they have fewer emergency department visits.
As described in Table 4, zolpidem’s AUC and Cmax kinetic values are 25% higher in females, and female patient counts surpassed males in the emergency department data for each of the 8 reported years from 2004 to 2011. In contrast, buprenorphine’s AUC and Cmax kinetic values were also higher in females (35%), yet male emergency department patients still exceeded females over the last five years of reporting (2007–2011). Do sex-dependent bioequivalence differences in Cmax or AUCs correlate with adverse events associated with emergency department visits?
Estimated emergency department visits related to buprenorphine: 2004 to 2011 DAWN data.

Anderson (2008), Rademaker (2001) and Bergiannaki and Kostaras (2016) have highlighted the fact that females are at higher risk for clinically relevant adverse drug reactions. They suggest that differences in pharmacokinetics, pharmacodynamics, and the actual dose of drugs, when expressed in mg of drug per kg of bodyweight are all contributing factors in the sex-based differential presentation of adverse events.
Generally, males weigh more than females, yet few drugs are dosed based on body weight. Plasma drug concentrations are dependent on the volume of distribution and clearance rates which are correlated with body weight for most drugs, independent of sex differences. Anderson (2008) also points to the fact that females have a higher percentage of body fat when compared to age-matched male cohorts which, in turn, affects the volume of distributions of certain drugs. Renal clearance of unchanged drug is also decreased in females due to a lower glomerular filtration rate. Rademaker (2001) suggests that because women generally have a lower lean body mass than males, they have reduced hepatic clearance, and have differences in activity of cytochrome P450 (CYP) iso-enzymes, as well. Rademaker reported that women had a 40% increase in CYP3A4, varied decrease in CYP2D6, and CYP1A2, and metabolize drugs at different rates compared with men. Bergiannaki and Kostaras (2016) concluded that women tend to have greater bioavailability and slower elimination of drugs leading to higher concentrations of free circulating drugs in serum and causing more side effects and adverse reactions to the psychotropic medication when compared to males. However, D'Incau et al. (2014) reported no sex-differences in the number of adverse events related to psychotropic medications in three European countries (France, Spain and Italy) when the data are normalized by population. For example, antidepressants initiated most reported adverse events (women, 303; men, 141; P < 0.001) but the reporting rates (number of adverse events reported divided by exposed patients in the same period) for these drugs, however, were not significantly different between women (0.87 cases per 10,000 treated persons per year) and men (0.81 cases per 10,000 treated persons per year).
With respect to sex differences in activity of the hepatic cytochrome P450 (CYP) and uridine diphosphate glucuronosyltransferase (UGT) enzymes that will result in differences in clearance rates, there is evidence for females having lower activity of CYP1A2, CYP2E1, and UGT; higher activity of CYP3A4, CYP2A6, and CYP2B6; and no differences in CPY2C9 and CYP2D6 activity. As cited by Moody et al. (2011) unlike the rat, no sex-specific drug metabolizing enzymes have been identified (Waxman and Holloway, 2009) in humans that can explain the imbalance between kinetics and associated adverse events. Other differences including bodyweight and composition have been cited for the disparity between pharmacokinetics and adverse event reporting in the emergency department DAWN data from SAMHSA.
Drug abuse liability test #1: Drug discrimination
The method by which a drug is administered to the animal is generally presumed to establish the rate and amount of a drug reaching the site of action. In designing two of the three core DAL tests described in the 2017 FDA guidance document, the pharmacokinetic data from early toxicology studies are useful but not required. Drug discrimination studies are conducted to assess the relative similarities of the internal, or subjective states induced by a new drug and those of a comparative known drug-of-abuse already scheduled under the CSA. For example, a new drug targeting dopamine for the treatment of Parkinson’s disease may administer the drug to rats that have been trained to discriminate the presence and absence of cocaine or amphetamine in the standard 2-choice lever press operant task under a fixed-ratio 10 schedule of food deliveries. The objective of this test is to answer one simple question, “is the new drug subjectively similar to cocaine or amphetamine in this well-established, FDA-acceptable and valid model of DAL?” For drug schedule control actions, it does not matter if the similarity in subjective effects occurs at 1.0 mg/kg, 10 mg/kg, or 100 mg/kg. If a new drug initiates “training drug-like” responding, (i.e. greater than 80% of the total lever press responses emitted on the cocaine or amphetamine lever in this assay), then it is considered to have some relative abuse potential, the specific dose of cocaine or amphetamine that engenders the shift is irrelevant to drug scheduling decisions.
In this example, a series of studies to assess the relative subjective effects of a new CNS active drug being investigated for the treatment of neuropathic pain is shown. The drug had a novel mechanism of action and the drug development team, in consultation with FDA reviewers, conducted drug discrimination studies using cocaine and morphine positive comparators. The intended route of administration of the test article was by oral administration, so discriminative cues were established through standard drug discrimination training procedures using the standard IP route for cocaine training and SC route for morphine stimulus control. Following successful training and generalization tests with the training cue route-of-administration, rats were tested the oral route for the training drugs.
As shown in Figure 1(a) and (b) above, rats trained to discriminate the presence versus absence of cocaine engendered similar patterns of cocaine-appropriate responding when tested with either IP or oral doses of the training drug (cocaine). Minor differences between males and females occurred along the ascending limb of the dose-response function, ranging from exclusive saline-appropriate responding at the low end of the dose continuum to exclusive cocaine-appropriate responding at the highest tested doses for both administration routes (refer to Craft and Stratmann, 1996; Anderson and van Haaren, 1999, 2000). Figure 1(b) shows the cross-generalization functions generated following both IP and oral dosing of a novel CNS-active test article conducted per the FDA abuse liability guidelines (2017). The test article engendered less than 20% cocaine-appropriate responding in both males and female, regardless of administration route. While there are slight differences in the percentage of total session responses emitted during each test session on the cocaine-appropriate lever, these differences have no functional significance with respect to a determination of discriminative accuracy or any of the 8-factors determinative of schedule control actions for this test article.

(a) Cocaine (CO) dose-response functions (DRF) for male (32 M, left panel) and female (32 F, right panel)) rats trained in a 2-choice cocaine (10 mg/kg, IP) versus saline drug discrimination assay under a FR-10 schedule of food deliveries in daily 30 min sessions with 15 min pretreatment intervals. Test sessions were identical to training sessions except for the injection administered prior to the sessions and rats were reinforced for 10-consecutive lever presses on either lever. DRFs were generated following intraperitoneal and oral gavage dose administrations of cocaine. (b) Cocaine (CO)cross generalization dose-response functions (DRF) for male (left panel) and female (right panel)) rats trained in a 2-choice cocaine (10 mg/kg, IP) versus saline drug discrimination under a FR-10 schedule of food deliveries in daily 30 min sessions with 15 min pretreatment intervals (see Figure 1(a)). Test sessions were conducted with a new CNS active test article under identical testing sessions for cocaine. Each selected dose of vehicle and test article were tested in 6 randomly selected male (left panel) and 6 randomly selected female (right panel) rats from the available pool of 32 trained rats shown in Figure 1(a). Subgroups were used to diminish any possible carry-over effects for test article administration cross-generalization profiles conducted by both IP and oral routes of administration. At minimum of one-week washout was imposed between any test.
Similar to the differential shape and distribution of the cocaine dose-response functions (Figure 1(a)) the morphine dose response functions showed sex-based differences in the morphine and test article dose response functions when conducted by either oral or subcutaneous routes of administration morphine trained rats (Figure 2(a) and (b)). The sex-based differences in the cross-generalization functions with a morphine training cue are comment-worthy but there are no functional or regulatory consequences to these sex-based differences in determining schedule control status of the new drug (refer to Craft et al., 1996; Craft, 2008) With respect to schedule control decisions under the CSA, the demonstration of cross-generalization or failure to show cross-generalization using two controlled substance comparators (morphine and cocaine) in both male and female rats represent an unnecessary use of animals and is inconsistent with the legal requirements of the Animal Welfare Act to reduce the number of animals used in research.

(a) Morphine (MO)dose-response functions (DRF) for male (32 M, left panel) and female (32 F, right panel)) rats trained in a 2-choice morphine (3.2 mg/kg, SC) versus saline drug discrimination assay under a FR-10 schedule of food deliveries in daily 30 min sessions with 15 min pretreatment intervals. Test sessions were identical to training sessions except for the injection administered prior to the sessions and rats were reinforced for 10-consecutive lever presses on either lever. DRFs were generated following subcutaneously and oral gavage dose administrations of morphine. (b) Morphine (MO) cross generalization dose-response functions (DRF) for male (left panel) and female (right panel)) rats trained In a 2-choice morphine (3.2 mg/kg, SC) versus saline drug discrimination under a FR-10 schedule of food deliveries in daily 30 min sessions with 15 min pretreatment intervals (see Figure 2(a)). Test sessions were conducted with a new CNS active test article under identical testing sessions for morphine. Each selected dose of vehicle and test article were tested in 6 randomly selected male (left panel) and 6 randomly selected female (right panel) rats from the available pool of 32 trained rats shown in Figure 2(a). Subgroups were used to diminish any possible carry-over effects for test article administration cross-generalization profiles conducted by both SC and oral routes of administration. At minimum of one-week washout was imposed between any test.
Drug abuse liability test #2: Self-administration
With almost 100 years of the scientific analysis of drug abuse in humans it was the early identification that consumers of alcohol and licit and illicit opiates that became physically dependent on the drugs could be returned to a relatively drug-free state through controlled or forced abstinence from the drug. However, the critical and more frustrating reports in drug abuse research was the almost universal finding that the majority of those “clean patients” would voluntarily return to consuming their drug of choice well after the disappearance of the physical signs of withdrawal (detoxification) and in full awareness of the destructive nature of their prior consummatory patterns of intake (Himmelsbach, 1942; Krueger et al., 1941–1943; Tatum et al., 1929). In 2019 the factors controlling such destructive cyclic behaviors remain elusive, but the importance of environmental conditions under which the drug is initiated, the environmental conditions related to drug withdrawal, and the environmental conditions under which the “drug clean” patient re-initiates drug administration remain the focus of addiction theories (McCrady and Epstein, 2013; Nyswander, 1956; Wikler, 1953).
Ator and Griffiths (2003) reviewed the positive correlation between those drugs that have been abused by humans and those drugs that animals self-administer is clear for opioids, stimulants, sedatives, dissociative anesthetics, and others. Over 40 years ago Griffiths and Balster (1979) summarized data that confirmed a strong correlation between opioids that were self-administered by monkeys and those that produced morphine-like signs, symptoms, and subjective-effects in humans. To say that a drug is “self-administered” in animals says as much about the experimental, drug, and environmental contingencies in effect at the time of dose deliveries as it does about the drug itself. The international drug control policies have been established based on a ranking of degrees of control relative to the potential or actual threat to human health and safety posed by the drug substance. Ator and Griffiths (2003) have also provided a definition for differentiating relative reinforcing efficacy of drugs which incorporates an important qualifier; one drug may be considered more reinforcing than another if it maintains self-administration under a wider range of conditions of availability, across laboratories, and species (Griffiths et al., 1979; Johanson et al., 1987; Schuster and Thompson, 1969). Using this behavioral “litmus test”, cocaine is an excellent example of a compound with high relative reinforcing efficacy. Cocaine (Schedule II, psychomotor stimulant) has a robust history as an effective reinforcer in training a variety of responses (lever presses, nose-pokes, etc.) in various laboratory animals, and will also maintain that responding under diverse conditions, including those with extremely high behavioral demands (Johanson and Fischman, 1989).
One critical determinative factor of schedule control of behavior lingering from the early learning paradigms is the principle that a stimulus that could serve as a robust primary reinforcer such as food, water, or a punisher such as electric shock, was determined by the latency between the completion of the operant demand and the delivery of that reinforcing or punishing stimulus (Azrin, 1960; Hake et al., 1967). Early learning theory proposed that response acquisition is negatively related to the interval between a response and its effect (Lattal, 2010; Skinner, 1938). Under the learning paradigm, intravenous drug administration as a stimulus event could be the reason why stimulus control of behavior by drugs had such a profound influence on human behavior, and why drugs may be the most robust reinforcing stimulus that factors into the long history of human drug abuse.
With the early use of food and water deliveries as reinforcers in standard learning assays the delay in reinforcer deliveries was a key feature in understanding the strength of stimulus control and reinforcer efficacy. However, the relative interoceptive/subjective effects induced by drug delivery to an animal in a drug self-administration study does not support the over generalization of the “delay of reinforcement” principle to CNS active compounds, like cocaine.
The 1996 labelling of the extended release formulation of oxycodone, OxyContin®, that was approved by the FDA included the following statement: Delayed absorption as provided by OxyContin tablets, is believed to reduce the abuse liability of a drug.
As it related to the reinforcing effects of a Schedule II opiate, this statement was inaccurate at the time. The stimulus event of drug delivery to the CNS is simply not a primary principle in human drug abuse patterns. As stated, above, young “psychonauts” experimenting with the neurotoxic agent 3,4 methylenedioxymethamphetamine (Ecstasy) prefer oral administration of the drug that is associated with a Tmax of 4 to 6 hours to achieve maximal hallucinogenic (entactogenic) effects. The reinforcing attributes of human growth hormone and anabolic steroid administration as performance enhancing drugs are certainly delayed from initial dose consumption.
Following oral administration, oxycodone is metabolized be the liver to the active minor metabolite oxymorphone by CYP2D6, and CYP3A4 catalyzes the drug to the major circulating metabolite (80%) noroxycodone. A third metabolite is noroxymorphone, derived from the metabolism of noroxycodone via CYP2D6 and to a lesser extent from oxymorphone via CYP3A4 (Graziania and Nisticò, 2016). Oral administration of a 20 mg dose of OxyContin does not have any significant differences between women and men with respect to the area under the curve (AUC) and in peak plasma concentration (Cmax) related to the oxycodone content of OxyContin, whereas the oxymorphone AUC was significantly greater in men compared with women. History soon provided further evidence that the “depot matrix system” (Acrocontin™) used to formulate high potency oxycodone into an extended release tablet initially containing a variety of doses from 10 to 160 mg of pharmaceutical grade oxycodone per tablet created a pharmaceutical nightmare for law enforcement and health care providers. Heroin abusers long tolerant to the “rush” of IV dosing needed only the long-term slow release attributes of oxycodone to ensure a stable state of plasma concentrations that would reduce the anxiety of an impending discontinuation syndrome to mark Oxycontin as the
For example, previous published reports have demonstrated that the rate of intravenous drug delivery in a standard operant self-administration assay may be varied from immediate to 40 seconds without any significant influence on the assessment of reinforcing effects (Balster and Schuster, 1973; Crombag et al., 2008; Panlilio et al., 1998). Other studies have shown that when infusions of cocaine or amphetamine were administered between 5 and 100 seconds, there was little-to-no effect on the acquisition or maintenance of self-administration behavior, progressive ratio breakpoints, or the reinstatement of drug-seeking behaviors (Kato et al., 1987; Liu et al., 2005; Pickens et al., 1969; Wakabayashi et al., 2010; Woolverton and Wang, 2004). Additionally, Samaha et al. (2011) have shown that conditioned reinforcing effects of drug cues are not critically dependent on the rate of dose administrations in rats using 5 second versus 90 second delivery intervals. The early conceptualization of the temporal relationship between stimulus and effects do not have robust support with such potent stimuli as CNS-active drug administrations. The effectiveness of drugs as reinforcers is demonstrated by the expenditure of work to gain delivery as well as the willingness to tolerate a delay.
Recently we completed a self-administration study in both male (32 M) and female (32 F) Sprague-Dawley rats under the conditions described in the FDA guidance document. Rats were conditioned in a standard single-lever operant task under a FR10 schedule of lever-press responding for the IV delivery of 0.56 mg/kg/injection of cocaine in daily one-hour sessions. To diminish the magnitude of a pharmacodynamic adaptation to repeated cocaine injections over the training period each rat was limited to 10 injections of cocaine in a single training session. Once day-to-day stability of voluntary intakes was demonstrated for each rat, test sessions were initiated under unlimited drug access conditions for 3 consecutive days of substitution for the maintenance dose of cocaine. Every three-session test series was preceded by the demonstration of a stable cocaine baseline performance for each rat. Complete dose response functions were generated in both male and female rats in ascending order under unlimited access conditions.
Sex-based differences in the incentive and motivational properties serving self-administration of cocaine in the rat have been reported previously by Lynch and Carroll (1999, 2000) and Kawa and Robinson (2019). While there are slight visible differences in the shapes and daily distributions of the dose-effect functions in our study for the total number of injections of cocaine self-administered in rats under unlimited access conditions, the reality of the data show that the group mean injections earned (34.7 ± 2.7 injections by males; and 35.1 ± 2.4 injections for females) and group means of total cocaine intakes from the maintenance dose of 0.56 mg/kg/injections (19.45 ± 1.5 mg/kg by males; 19.64 ± 1.3 mg/kg by females). The intravenous threshold dose of cocaine to induce seizures in rats has been reported to be approximately 20 mg/kg.
As shown in Figure 3(a) and (b), if rats trained to consistently lever press for delivery of an intravenous bolus of cocaine, continued to lever press and self-deliver doses of a new drug over 3 to 5 consecutive days of testing, then the new drug is considered to have abuse liability. The self-administration assay is simply asking one question, will the new drug initiate and maintain operant responding for its own delivery in rats that have been conditioned to self-administer a known drug of abuse, like cocaine? If a new drug maintains stable day-to-day intakes when tested under unlimited access conditions under a fixed-ratio 10 schedule of drug deliveries (as described in the FDA guidance document), the drug is assumed to have a abuse liability potential for human consumers once approved for marketing. Both males and females self-administer opiates – the specific threshold or preferred doses of male or female rats are irrelevant to schedule control actions.

(a) Dose response functions for 32 male (left panel) and 32 female (right panel) Sprague-Dawley rats conditioned to self-administer a maintenance dose of 0.56 mg/kg/injection of cocaine (maintenance dose; MD) in a single-lever operant task under a fixed ratio 10 schedule of drug deliveries in daily one-hour sessions with a maximum of 10 injections per day during conditioning. Once stable daily intakes of the maintenance dose of cocaine was achieved for each rat on study, defined as less than 20% variability in voluntary injections for three consecutive days, substitution test sessions were conducted. All tests were conducted under unlimited access conditions. Saline and 0.56 mg/kg/injection were tested in all rats. Other doses were tested for 3 consecutive days under unlimited access conditions in 6 randomly selected male (left panel) and female (right panels). (b) Total amount of cocaine self-administered in male (left panel) and female (right panel) Sprague-Dawley rats during 3 consecutive daily one-hour substitution test sessions. Each bar for test doses of 0.1, 0.18, 0.32, and 1.0 mg/kg/injection of cocaine represents the group mean of 6 rats. 0.56 mg/kg/injection of cocaine and saline were tested for 3 consecutive days in 32 M and 32 F rats. Each open symbol (dash-dot line) represents the 3-day grand mean for each testing condition.
Drug abuse liability test #3: Dependence potential
The third abuse liability behavioral assay described in the FDA guidance document and recommended to be conducted in laboratory purpose-bred rodents has a minimal impact on the decision to initiate schedule control actions by either DEA or FDA. The potential to induce a state of physiological dependence, defined by the expression of discontinuation or withdrawal following abrupt cessation of treatment during a standard 15 to 30-day repeat dosing study design. A unitary positive study finding in the DAL testing a schedule control review would not be initiated. The identification or verification of the dependence liability of a new CNS active drug is more critical for labelling purposes. Many legend drugs like corticosteroids, propranolol, and serotonin-selective uptake inhibitors (SSRIs) induce a discontinuation syndrome if treatments are abruptly stopped, but none of these drugs are schedule controlled. To identify that females have a more pronounced discontinuation syndrome when compared to males, is not relevant for schedule control placement. If both males and females show signs of any withdrawal it is considered a predictive factor for the potential for abuse by humans once the NDA is approved.
The bioequivalence data discussed above regarding sex-based PK differences in Cmax and AUCs are extremely relevant to the expression of drug withdrawal and the differential severity of discontinuation syndromes experienced between men and women. The vast set of data regarding the experimental induction of drug dependence and the subsequent intensity of the discontinuation syndrome quantified under laboratory-controlled conditions in both animals and humans has clearly demonstrated that long-acting drugs are less likely to produce physical dependence when compared to short-acting CNS drugs. Wulff (1959) has been attributed in defining the importance of the rate of drug elimination in the overt expression of drug withdrawal symptoms. No withdrawal symptoms were observed by Wulff in patients administered long-acting barbital or pentobarbital. However, severe signs of convulsions and delirium were observed following the cessation of treatment of the short-acting barbiturates, like methohexital. For females with higher concentrations of CYP3A4 the elimination rate for barbiturates is faster than male cohorts. With respect to CNS depressants, like barbiturates, Okimoto (1977) reported that the most dramatic effect of increasing the rate of elimination by sex-based metabolic enzyme content of the liver is the greater incidence and magnitude of convulsions, death, and the total number of signs of withdrawal in females when compared to males. However, even with these health consequences from abrupt cessation of treatments, the dependence potential of any new CNS active drug does not, on its own, initiate drug control scheduling actions.
Table 5 lists some of the most severe signs of the discontinuation of chronic CNS depressant dosing Drug withdrawal of the barbiturate-type is severe and must be conducted under medical supervision.
Sex based differences in the expression of drug withdrawal of the barbiturate-type attributed to differential CYP3A4 hepatic concentrations.

As described above, opiates are metabolized by both CYP2D6 and CYP3A4 microsomal enzymes. While there are racial (genetic) differences in the expression of CYP2D6, CYP3A4 has been shown to have a strong chromosomal sex-based differential expression that affects the elimination rates of opiates between males and females.
Multiple factors are involved in selecting the doses for dependence liability studies. According to Okamoto (1984) and Hollister (1980) drugs which have long half-lives are less likely to result in severe withdrawal signs and symptoms despite the fact that they produce severe physical dependence. That is, the CNS can gradually re-adapt, when the drug is no longer administered, due to the slow elimination of the drug from the CNS. Additionally, the dose of drugs must be selected as to ensure that the CNS is chronically exposed to the drug during the dosing interval. As drug plasma concentrations approach zero, withdrawal reactions will be expressed. If the half-life of the compound is short, then the animal will be exposed to many small episodes of withdrawal over the course of dosing.
In Title 21, Chapter 13, §811(c)(7), the U.S. Congress has required that both the psychic and physical dependence liability of new chemical entities must be fully assessed relative to drugs listed in the Schedule targeted for placement of the new chemical entity. Similarly, the WHO Technical Report (#903), titled, “WHO Expert Committee on Drug Dependence (Geneva, 2001) has defined the criteria for control under International Treaties to include, the substance has the capacity to produce (a) a state of dependence, and (b) central nervous system stimulation or depression, resulting in hallucinations or disturbances in motor function, thinking, behaviour, perception, or mood; or the substance has the capacity to produce similar abuse and similar ill effects to a substance in Schedule I, II, III, or IV.
The FOB data above show the degree, magnitude or intensity, and duration of the withdrawal syndrome from morphine in both male and female rats. The temporal patterns of changes in the individual parameters of the 5 clustered domains over 15 days following abrupt cessation of twice daily dosing (terminal 150 mg/kg, bid morphine) represented a similar withdrawal syndrome of the opiate-type in both male and female rats. There were no statistical or physiologically-relevant differences (quantitative or qualitative) in a majority of the standard FOB parameters measured once a day over the 15 day post-treatment drug withdrawal assessment interval, including:
Activity Arousal Domain: Handling reactivity and vocalization scores. Autonomic Domain: salivation, lacrimation, pupil response to light, piloerection, exopthalmus scores; Neuromuscular Domain: Forelimb and Hindlimb grip strength, hindlimb splay, stereotypy score, tonic and clonic movements, and mobility scores. Sensorimotor Domain: responses to approach, click and touch; Physiological Domain: respiratory function.
While some quantitative differences between male and female rats can be observed (see Figures 4 to 8), a conclusion of a prototypic drug dependence of the opiate type can be assessed in either male or female subjects; both sexes are not needed.

Individual parameters from male (left panels) and female (right panels) rats summarized on a hypothesized activity/arousal domain assessed using repeated functional observational batteries conducted daily for 15 days following abrupt cessation of twice daily treatments of high dose morphine administrations or saline in laboratory rats. Subjective ordinal scale measurements were conducted for “ease of removal” and “arousal scores”, however, actual rearing counts during 3 min open field assessments were documented by independent raters, blind to treatments. Withdrawal of the opiate-type is generally expressed within the first week of “recovery”.

Individual parameters from male (left panels) and female (right panels) rats summarized on a hypothesized autonomic domain assessed using repeated functional observational batteries conducted daily for 15 days following abrupt cessation of twice daily treatments of high dose morphine administrations or saline in laboratory rats. Subjective ordinal scale measurements were conducted by independent raters, blind to treatments. Withdrawal of the opiate-type is generally expressed within the first week of “recovery”.

Individual parameter from male (left panels) and female (right panels) rats summarized on a hypothesized neuromuscular domain assessed using repeated functional observational batteries conducted daily for 15 days following abrupt cessation of twice daily treatments of high dose morphine administrations or saline in laboratory rats. Subjective ordinal scale measurements were conducted by independent raters, blind to treatments. Withdrawal of the opiate-type is generally expressed within the first week of “recovery”.
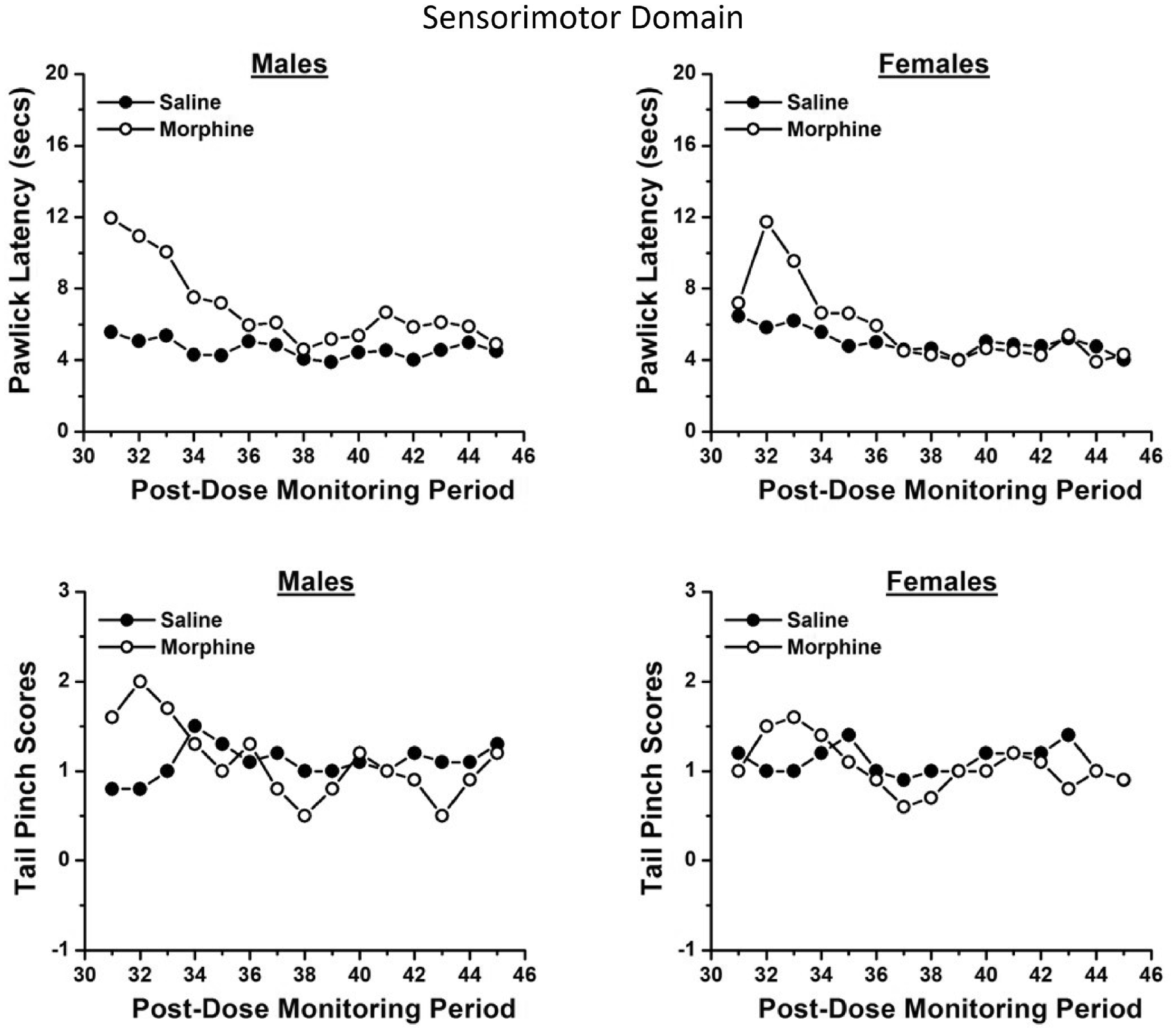
Individual parameter from male (left panels) and female (right panels) rats summarized on a hypothesized sensorimotor domain assessed using repeated functional observational batteries conducted daily for 15 days following abrupt cessation of twice daily treatments of high dose morphine administrations or saline in laboratory rats. Subjective ordinal scale measurements were conducted for reactivity to a tail pinch and actual time of an identified escape response (paw lick latency, or jumping,etc.) as a measure of analgesia was recorded by independent raters, blind to treatments. Withdrawal of the opiate-type is generally expressed within the first week of “recovery”.

Individual parameter from male (left panels) and female (right panels) rats summarized on a hypothesized physiologic domain assessed using repeated functional observational batteries conducted daily for 15 days following abrupt cessation of twice daily treatments of high dose morphine administrations or saline in laboratory rats. Quantified measures of body weight and body temperatures were recorded by independent raters, blind to treatments. Withdrawal of the opiate-type is generally expressed within the first week of “recovery”.
In rodents, the discontinuation syndrome expressed following high dose repeat administration of opiates is characterized by a constellation of somatic signs, such as jumping, paw tremor, wet dog shakes, and body stretch, which are mediated by multiple diffuse sites in the brain (Bozarth and Wise, 1984; Gellert and Holtzman, 1978; Gold et al., 1994; Maldonado et al., 1992; Wei et al., 1973). Although quantitative sex differences can be detected, -sex related qualitative differences have the most valuable implications.
Some researchers have employed opioid receptor antagonist-precipitated opiate withdrawal procedures (Linseman, 1977; Mucha et al., 1979; Schulteis et al., 1994). Papaleo and Contarino (2006) have concluded that opioid antagonists increase the intensity, change the timing and the frequency of most of opiate withdrawal signs and may trigger behaviors, such as writhing, hostility on handling, ptosis, rhinorrhea, lacrimation, and penile ejaculations that are usually absent in animals undergoing spontaneous opiate withdrawal (Linseman, 1977; Mucha et al., 1979; Ruiz et al., 1996). As summarized in Table 6, spontaneous and opioid receptor antagonist-precipitated opiate withdrawal conditions also differentially affect the activity of endogenous opioid systems in several brain regions.
Differences in brain biochemistry and behavioral expression of withdrawal of the opiate type using spontaneous cessation of treatments or naloxone-injections to precipitate the withdrawal.

Differences in the expression of opiate withdrawal reported in the published literature may be the result of the two methods used to characterize the syndrome. The abrupt cessation of opiate dosing to allow for “natural” withdrawal to occur will not necessarily show identical signs of withdrawal when compared to naloxone-precipitated opiate withdrawal. The opiate antagonists are not benign substances without CNS effects of their own. Opiate receptor antagonists may mask gender- and drug-linked differences in the expression of somatic opiate withdrawal that are instead observed in rodents undergoing spontaneous opiate withdrawal.
Conclusions
Non-clinical abuse liability testing is required for all CNS active compounds. The current thinking of the US FDA (2017) is expressed in the finalized abuse liability guidelines. The FDA clearly states that the presence of physical dependence or tolerance does not determine whether a drug has abuse potential. For the FDA, the primary focus is on whether a drug has rewarding properties, the ability of that drug to induce physical dependence or tolerance may influence its overall abuse potential. In determining whether an expensive human abuse potential study should be conducted, the FDA has expressed interest in determining if the new drug produces any of the following:
Responses in animals in general behavioral studies that are similar to responses to known drugs of abuse; Generalization (similar effects) to a known drug of abuse in animal drug discrimination studies; and Rewarding properties that support animal self-administration or conditioned place preference;
To date, drug scheduling of new drugs by the DEA has never placed differential schedule controls on a drug based on sex-based drug sensitivity (PK/PD). While there are sex-based differences in the degree and magnitude of adverse events associated with drug use outside the scope of medical practice (Emergency Room data and Treatment Episode data), and sex-based differences in the “preferred drug of choice” among different pharmacological classes of common drugs-of-abuse, all current International and federal drug scheduling has been “sex-neutral”. As a matter of record, the FDA reviewed 300 new drug applications between 1995 and 2000. Of the 163 that included a sex analysis, 11 drugs showed a > 40% difference in pharmacokinetics between males and females, which was listed on the product label, yet not a single dosing recommendation was made based on sex (Anderson, 2005).
Based on the administrative histories and NDA approval pharmacological reviews available on the FDA website the three behavioral assays qualified by the FDA for abuse liability testing for inclusion in NDA applications (drug discrimination, self-administration and dependence liability) have not shown a significant quantitative differential response that would meet the regulatory weight of evidence required to request differential schedule controls on any new drug approved over the last 30 years.
With respect to self-administration studies, FDA has given dispensation for the inclusion of both sexes if studies are conducted with NHPs. If sex-differences are a regulatory standard by which key decisions on drug control are based, what administrative policy is there to violate the Animal Welfare Act when using rats. Examples of new drugs where the sex specific data impacted the regulatory decisions seem appropriate. In a relatively recent review of the drug discrimination literature, Glennon and Young (2011) reported: 1) sex-dependent differences in the number of training sessions required to reach behavioral criteria for stimulus control by some drugs-of-abuse, 2) the potency of a training drug or positive comparator to engender training-drug appropriate responding in group mean data, and 3) the duration of action of a drug in the model. None of these dependent measures are used for abuse liability determinations in the 8-factor analysis determinative of schedule control (21 USC §811) by DEA. Findings across epidemiological studies have consistently demonstrated that both lifetime and 12-month prevalence alcohol and substance abuse disorder diagnoses are higher in males when compared to females (Epstein and Menges, 2013). Based on these finding what is the reasoning that drives continued testing of both sexes in abuse potential studies? Podolsky and Lukas (1999) reminds us all that governmental agencies cannot arbitrarily require additional demands, financial burden, or record keeping for regulated entities like pharmaceutical drug developer registrants. Administrative agencies such as the FDA are themselves regulated under the Administrative Procedures Act (APA: 5 USC, Part 1, Ch. 5) which requires them to publish their proposed requirements in the Federal Register and consider public comment before implementing new regulatory requirements.
Through the APA process, FDA has adopted the International Conference on Harmonisation Guidance M3(R2) on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (https://www.fda.gov/media/71542/download) as well as the S7A Safety Pharmacology Studies for Human Pharmaceuticals (https://www.fda.gov/media/72033/download). In adopting these Guidelines the FDA stated: This guidance is being issued consistent with FDA’s good guidance practices regulation (21 CFR 10.115). The guidance represents the agency’s current thinking on this topic. It does not create or confer any rights for or on any person and does not operate to bind FDA or the public. An alternative approach may be used if such approach satisfies the requirements of the applicable statutes and regulations.
The FDA’s adoption of the ICH M3(R2) and S7A may supersede the NIH’s sex-as-a-biological-variable (SABV) announcement from 1998.
McHugh et al. (2018) provided a 30 year review of the drug abuse literature since the NIH adoption of the SABV policy (1985). In their opinion, the trends in addiction medicine suggest that much of the historical gender differences in the prevalence of both substance use and SUDs may be attributable to social and cultural factors and not biological sex differences. The current total weight of evidence of nonclinical abuse liability testing predictive of human drug abuse does not support the need to conduct these tests in both male and female purpose-bred animals. Requests by CSS to include both sexes in these assays seem to be in direct opposition to the statutory intent of the Animal Welfare Act (1994b). To reduce the use of animals in research we suggest that the current literature supports the continued use of only the most pharmacologically sensitive sex to screen for abuse liability, not both.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.