
Abstract
Background:
Interferon regulatory factor-2 binding protein 2 (IRF2BP2) is an important new transcriptional cofactor that interacts with IFN regulatory factor 2 (IRF-2) and an IRF-2-dependent transcriptional repressor. IRF2BP2 plays a role in different cellular functions, including apoptosis, survival, and cell differentiation. In this study, we report a case with common variable immunodeficiency (CVID), which has a heterozygous variant in the IRF2BP2 gene.
Case Presentation:
A 13-year-old girl was evaluated for immunodeficiency due to recurrent sinusitis and tonsillitis in the previous year. She had been suffering from chronic cough for 3 months. She was hospitalized with lobar pneumonia and bronchiectasis. She was the second child of consanguineous parents. On physical examination, there was no growth and development retardation. Immunological screening of the patient demonstrated panhypogammaglobulinemia with low total memory B and class-switching memory B cells. Specific antibody responses to rubella and hepatitis B were negative. T- and B-lymphocyte counts and T-cell responses to phytohemagglutinin (PHA) were normal. Exome sequencing identified a heterozygous variant in IRF2BP2 (c.112C>Tp.Arg38Cys). On follow-up, she has maintained a good infection control with antibiotic prophylaxis and immunoglobulin replacement therapy.
Conclusion:
To the best of our knowledge, this case is the youngest CVID who was diagnosed with IRF2BP2 in the literature. The low percentage of total memory and switched memory B cells in the proband suggested that IRF2BP2 might have had a role in the development or survival of memory B cells. Functional studies are needed about the critical role of IRF2BP2 protein in B-cell maturation and humoral immune responses.
Introduction
Common variable immunodeficiency (CVID) is the most common symptomatic inborn error of immunity. 1 According to the European Society for Immunodeficiencies, the diagnostic criteria for CVID require the presence of symptoms and laboratory abnormalities including (1) at least one of after: increased susceptibility to infection, autoimmune manifestations, granulomatous disease, unexplained polyclonal lymphoproliferation or affected family member with antibody deficiency; (2) marked decrease of IgG and marked decrease of IgA with or without low IgM levels; and (3) at least one of the after: poor antibody response to vaccines or low switched memory B cells. Secondary causes of hypogammaglobulinemia must be excluded, there must be no evidence of profound T-cell deficiency, and the diagnosis should be established after the fourth year of life. 2
To date, a monogenic cause has been identified in only 10%–30% of patients with CVID.3,4 IFN regulatory factor-2 binding protein 2 (IRF2BP2), one of 20 genes associated with CVID phenotypes, has an unclear role in immune dysregulation, with a limited current understanding of its mechanisms and effects.3,4 IRF2BP2 is an important transcriptional cofactor. It binds at its C-terminus to IFN regulatory factor 2 (IRF-2) to act as a transcriptional corepressor. 5 Additionally, IRF-2-independent functions of IRF2BP2 may be indicated as a gene expression regulator—for example, as a repressor of nuclear factor of activated T-cell (NFAT)-mediated transcriptional activity (Fig. 1). IRF2BP2 acts as a corepressor of NFAT by inhibiting its transcriptional activity. Specifically, IRF2BP2 interacts with NFAT and prevents it from activating target gene expression, thereby modulating immune responses and inflammation. This regulatory function is of particular importance in the context of macrophage function, lymphocyte activation, and angiogenesis, which are critical processes in cancer, cardiovascular diseases, and immune regulation. By suppressing NFAT activity, IRF2BP2 helps to control excessive immune activation and inflammatory responses. 6 However, the precise mechanism by which IRF2BP2 pathogenic variants impair immune system functions remains unclear. 7
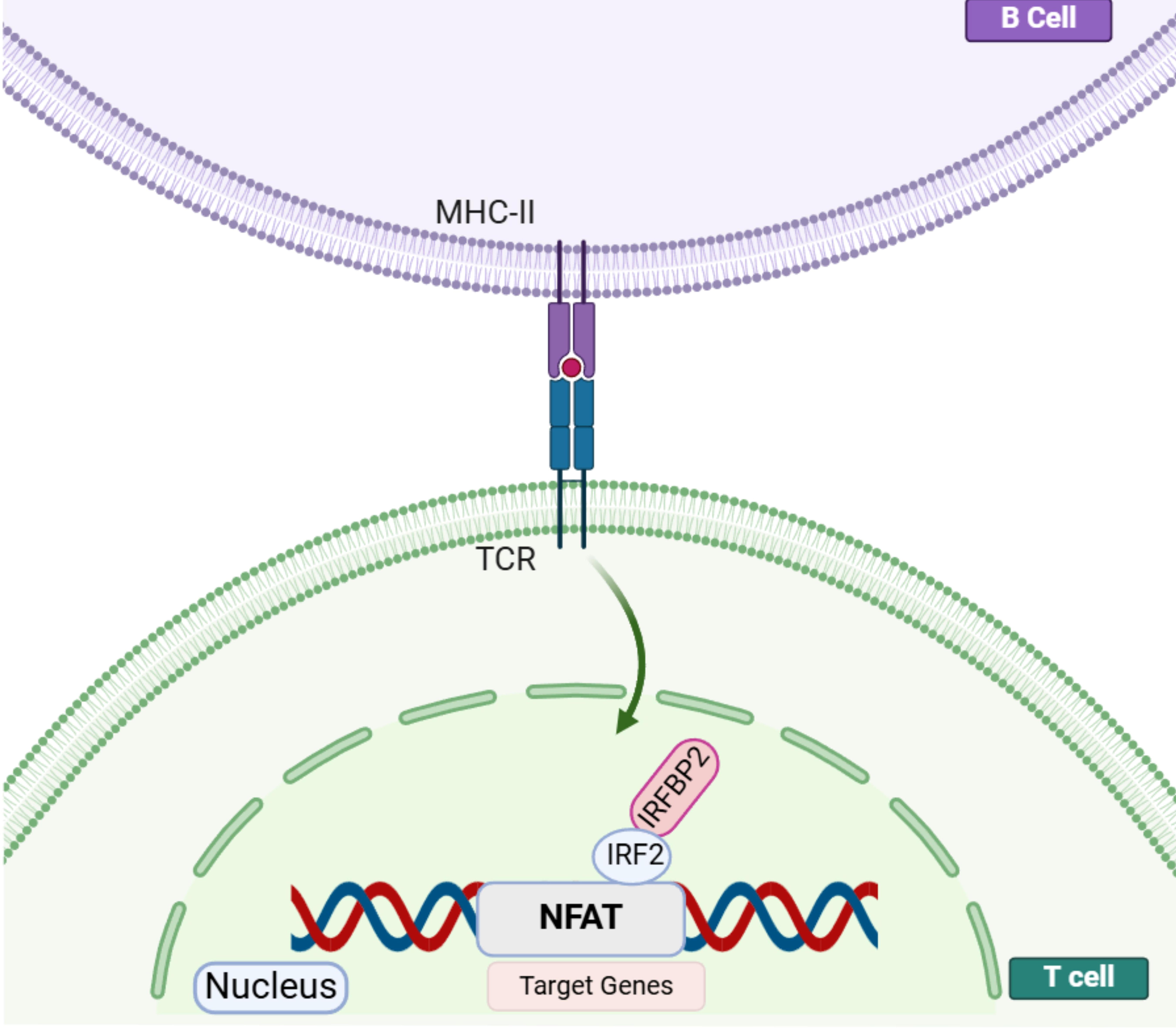
Drawn with BioRender. NFAT proteins enter the nucleus, bind to their target genes, and can activate or repress transcription. IRF2BP2 is a negative regulator of the NFAT-mediated transcriptional activity. IRF2BP2 acts as a co-repressor of IRF2 by binding to it through the C-terminal RING domain and is essential for IRF2 ability to limit the expression of IFNs. After T-cell activation by antigen presentation, NFAT, IRF2, and IRF2BP2 molecules colocalize in the nucleus. Expression of IRF2BP2 on CD4+ T cells leads to NFAT repression of target genes and decreased production of IL-2 and IL-4 in these cells. IL, interleukin; IRF2, IFN regulatory factor 2; IRF2BP2, interferon regulatory factor-2 binding protein 2; NFAT, nuclear factor of activated T cell.
This study presents an adolescent girl with CVID who has a heterozygous variant in the IRF2BP2 gene. This variant has rarely been described in the literature for pediatric patients.8,9
Case Presentation
A 13-year-old female was evaluated for immunodeficiency due to a history of recurrent sinusitis and tonsillitis over the past year, in addition to a chronic cough that had persisted for 3 months. Her medical records revealed no history of frequent illnesses prior to the age of 12 years. A physical examination revealed the presence of widespread crepitant rales, no dysmorphisms, and no evidence of growth or developmental delay. The patient was hospitalized for treatment due to the presence of lobar pneumonia and bronchiectasis detected on thorax tomography (Fig. 2). Her parents were second-degree cousins, and she had a healthy 15-year-old sister and no known family history of immunodeficiency.
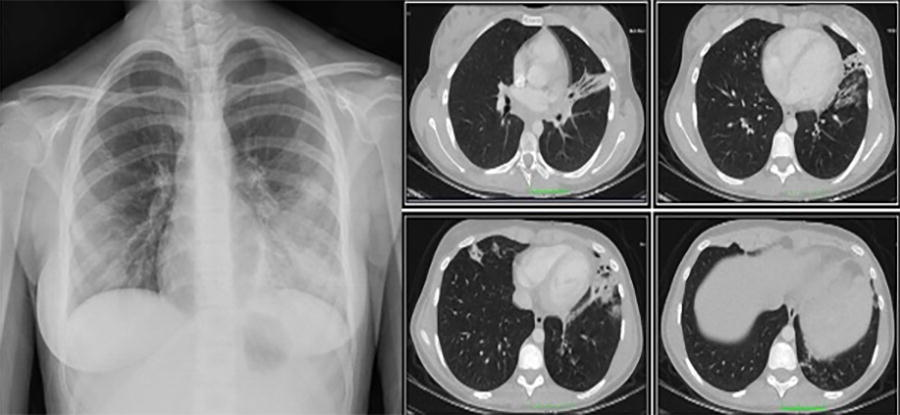
Chest X-ray and computed tomography scan showing bronchiectasis and bronchial wall thickening with left lower lobe predominance.
In the laboratory findings, total neutrophil and lymphocyte counts were within the normal ranges; however, serum immunoglobulin levels were significantly low: IgG < 143 mg/dL, IgA < 6.6 mg/dL, IgM at 14.6 mg/dL, and IgE < 19 mg/dL. Specific antibody responses to rubella and hepatitis B were negative (Table 1). The isohemagglutinin titer was normal.
Immunological Data of the Patient
IgRT, intravenous immunoglobulin replacement.
Regarding immunological screening, natural killer (NK) and B-cell counts were lower than the normal ranges, but T-cell ratios and responses to PHA were normal. B-cell subset immunophenotyping with flow cytometry revealed that she had very low total and switched memory B cells and normal CD19+CD21−/low B cells (Fig. 3). The NK cell ratio has reached to normal in the follow-up controls. The low count at the time of diagnosis was considered to be secondary to viral infection (Table 1).
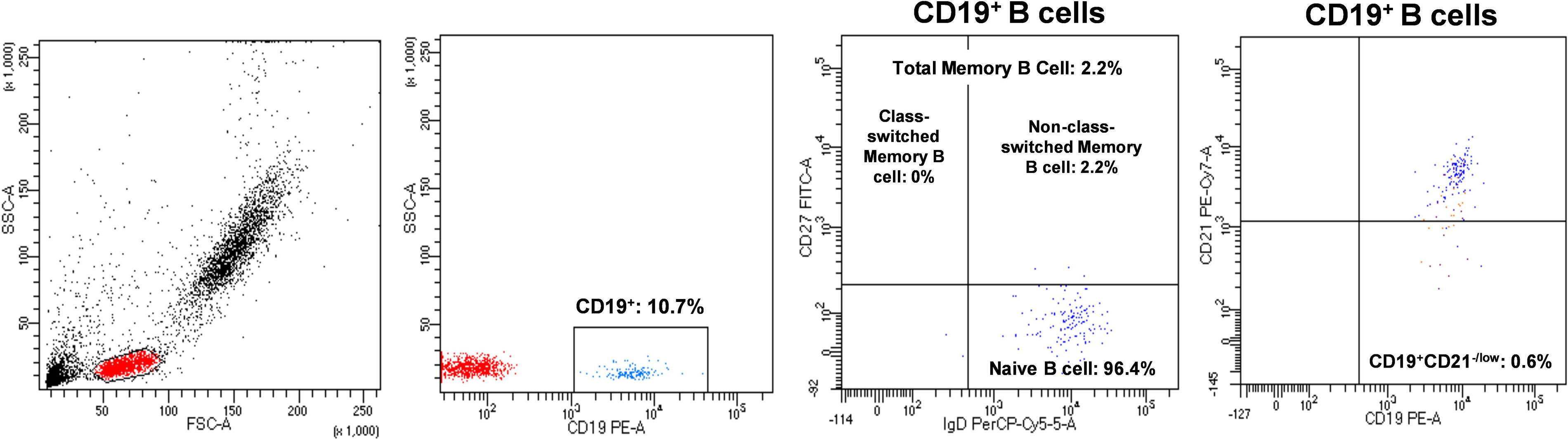
B-cell immunophenotyping in the IRF2BP2-deficient patient showing decreased total and switched memory B cells and normal CD19+CD21−/low B cells. IRF2BP2, interferon regulatory factor-2 binding protein 2.
Exome sequencing analysis indicated a heterozygous variant in the IRF2BP2 gene—NM_182972.3 c.112C > T (p.Arg38Cys; OMIM #617765), which is classified as a variant of uncertain significance according to the ClinVar, Franklin, and VarSome databases (Fig. 4). Hence, in-silico prediction tools were used to assess the potential impact of this variant, which was determined as deleterious, with the Combined Annotation Dependent Depletion (CADD) score: 28.7; PolyPhen-2 score: 0.999. It exhibits lower protein stability (ΔΔG 0.23). No IRF2BP2 variant was identified in the patient’s parents and sister.
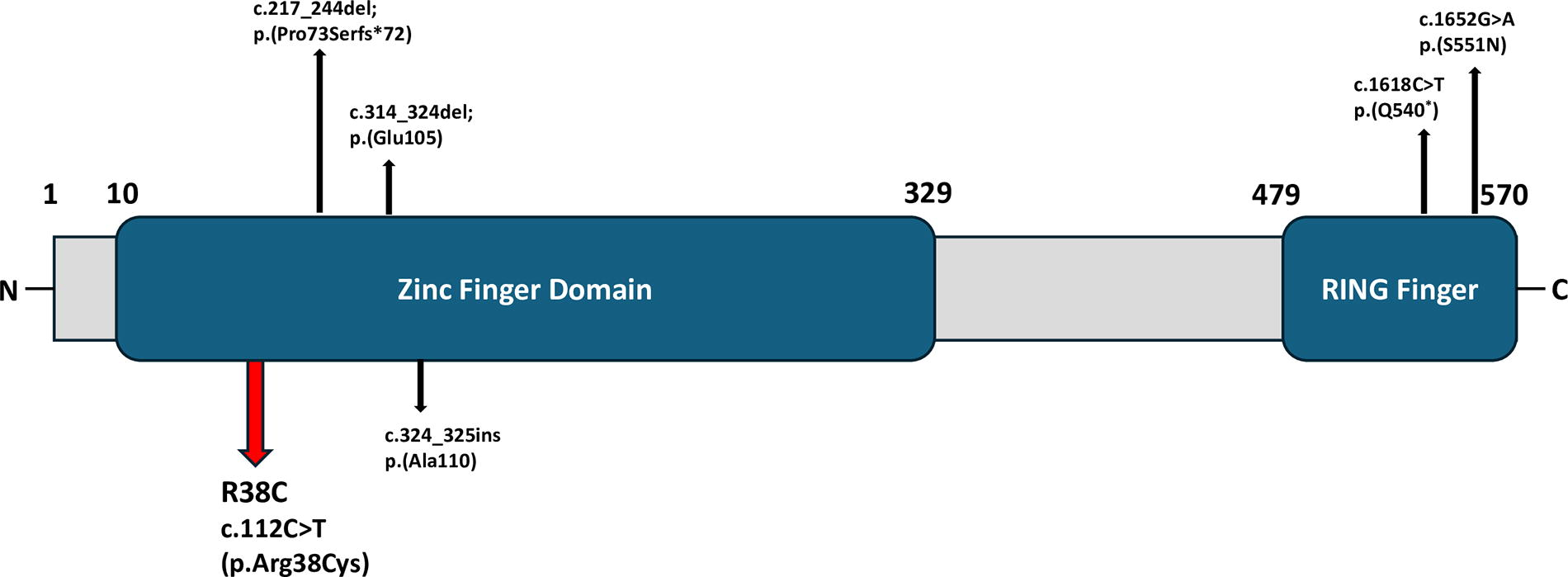
Schematic of the IRF2BP2 protein with a N-terminal zinc finger domain and a C-terminal RING domain. The variant in our patient is located in the zinc finger domain and is indicated by a red arrow. Black arrows indicate IRF2BP2 variants in the other patients with CVID published in the literature. CVID, common variable immunodeficiency; IRF2BP2, interferon regulatory factor-2 binding protein 2.
The patient, who has a history of recurrent sinopulmonary infections and bronchiectasis, panhypogammaglobulinemia, inadequate antibody responses to vaccines, and a low number of class-switched memory B cells, was diagnosed with CVID according to the criteria set forth by the European Society for Immunodeficiencies. 2 She was placed on trimethoprim–sulfamethoxazole (5 mg/kg/dose, PO, daily) for prophylaxis and on intravenous immunoglobulin replacement (IgRT, 0.5 g/kg/dose every 3–4 weeks). At the follow-up, it was observed that pancytopenia occurred secondary to acute viral infection (influenza B positive and Epstein–Barr Virus DNA negative) but resolved in 2 weeks. Despite the absence of complications over a 2-year period, during which the patient was treated with antibiotic prophylaxis and IgRT, she is being monitored for the potential development of autoimmune, lymphoproliferative, and malignant diseases that may arise in patients with CVID. Informed consent was obtained from the patient’s parents for publication.
Discussion
This report presents a pediatric patient with CVID who has an IRF2BP2 heterozygous variant—NM_182972.3 c.112C > T (p.Arg38Cys)—but no clinical case has been reported in the literature. Although IRF2BP2 deficiency has been identified among monogenic CVIDs, only a limited number of patients with missense and nonsense IRF2BP2 variants and an autosomal dominant inheritance pattern have been documented to date.8–10 Our patient had a heterozygous variant, and other family individuals do not carry the same variant, indicating pathogenic segregation of the variant. However, the potential for another intronic variant that might have an additional effect on pathogenicity could not be excluded. To our knowledge, considering Garzia’s latest study, 10 families with IRF2BP2 mutations have been described in the literature thus far. Consequently, most of these patients have been diagnosed with CVID. The patient presented here is the youngest with an IRF2BP2 mutation identified in the literature as having CVID.
The first reported family phenotypic spectrum was characterized by a CVID-like immunodeficiency with hypogammaglobulinemia and recurrent sinopulmonary infections, as well as autoimmune symptoms (such as psoriasis, colitis, and type 1 diabetes). All identified family members exhibited normal total B-cell counts. However, two patients displayed a relative decrease in the number of switched memory B cells, which is consistent with the findings observed in our patient through flow cytometry. 8
Baxter et al. later described a 4-year-old case with an IRF2BP2 heterozygous mutation compatible with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX). The patient had chronic diarrhea, severe eczema, anemia, failure to thrive, fevers, a short stature, recurrent infections, cataracts, hypodontia, alopecia, and hypogammaglobulinemia. There was no information about the patient’s flow cytometry results and B-cell subsets. 9
Pan et al. reported the case of a 22-year-old patient with a heterozygous mutation in IRF2BP2 who, at follow-up, presented with recurrent infections and autoimmune disorders (i.e., Crohn’s disease and autoimmune encephalitis). His immunoglobulin values before intravenous immunoglobulin (IVIG) treatment are unavailable, but there was a decrease in B-cell plasmablasts (CD19+CD27+CD38+). 10
Körholz et al. described a 33-year-old male patient with a de novo IRF2BP2 variant and a diagnosis of CVID. Since the age of 8 years, he had been receiving IgRT for recurrent lower respiratory tract infections and hypogammaglobulinemia. At the age of 25 years, he developed autoimmune symptoms such as colitis and rheumatoid arthritis. This case demonstrates the importance of early genetic diagnosis, especially in patients diagnosed with primary immunodeficiency in childhood. 11
In 2023, Garzia-Aznar et al. reported five novel loss-of-function mutations in IRF2BP2. The authors examined a total of five family members and observed that four patients were diagnosed with CVID. Three of these patients had bronchiectasis, similar to our patient. Another patient diagnosed with CVID later developed Crohn’s disease, which was identified at follow-up. Although the other three patients were diagnosed in adulthood, the patient diagnosed with CVID at the age of 15 had bronchiectasis, recurrent infections, hypogammaglobulinemia, a poor response to vaccines, and phenotypic features such as hypotonia, pectus carinatum, and hyperplexia. Information on the B- and T-cell immunophenotype of this patient is unavailable. 12 Again, this report presents another case with a 13-year-old male. He had a history of recurrent laryngitis and bronchopneumonia in early childhood, hemiplegia as a side effect of the varicella vaccine at the age of three, and hospitalization with chickenpox virus meningitis at the age of 13. However, unlike our patient, this patient had normal immunoglobulin values and was not diagnosed with CVID. 12
Autoimmunity affects up to 30% of patients with CVID and often serves as the initial manifestation of immunodeficiency at the disease’s onset.13,14 As observed in patients with IRF2BP2 genetic defects previously reported in the literature, they usually have at least one autoimmune symptom at the time of diagnosis and during follow-up. In 2024, Anim et al. described three rare novel variants in IRF2BP2, identified in patients with primary antibody deficiency and autoimmunity by whole-exome sequencing in their study. 15 The most notable aspect of previously documented patients was the presence of comorbidities such as diabetes, psoriasis, and colitis, which are associated with autoimmunity. Our case, under our observation for 2 years, has not manifested any of the aforementioned autoimmune symptoms. This may be attributable to her early diagnosis and relatively young age.
This case illustrates that IRFBP2 variant may also be responsible for cases of CVID developing in childhood. Further research is required to elucidate the pivotal function of IRF2BP2 in B-cell maturation and humoral immune responses.
Footnotes
Authors’ Contributions
All authors approved the final article as submitted and agree to be accountable for all aspects of the work.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
No funding was received for this article.